

The Ma’ayan Laboratory develops computational methods to study the complexity of regulatory networks in mammalian cells. We apply AI and other statistical mining techniques to study how intracellular regulatory systems function as networks to control cellular processes such as differentiation, dedifferentiation, apoptosis, and proliferation. We develop software systems to help experimental biologists form novel hypotheses from high-throughput data, while aiming to better understand the structure and function of regulatory networks in mammalian cellular and multi-cellular systems.
Browse through the following categories to explore the various original resources we have developed:
- Most Popular
- Recently Published
- Enrichment Analysis
- Drug and Target Discovery
- Computational Platforms and Workflow Engines
- Gene and Drug Pages
- lncRNAs
- Data and Information Portals
- Data Visualization Components
- Deprecated
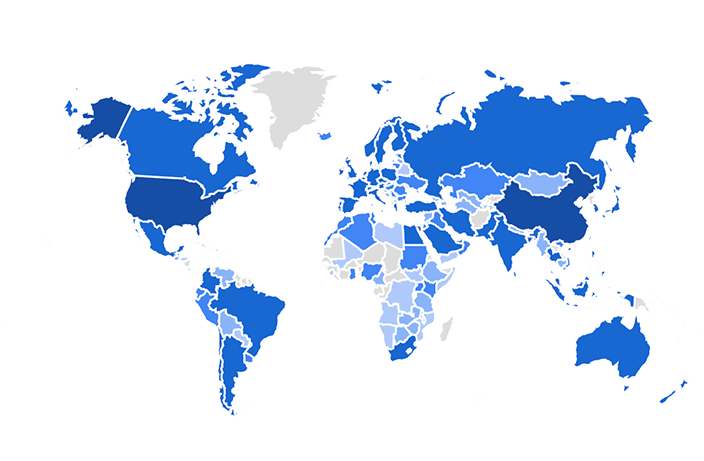
We apply these and other computational methods for the analysis of a variety of collaborative projects. The results from our analyses produce concrete suggestions and predictions for further functional experiments. The predictions are tested by our collaborators and our analyses methods are delivered as software tools and databases for the systems biology research community. So far, over three million unique users have accessed the web-based resources that we have developed. Below is a screenshot from Google Analytics that shows the distribution of users from countries that accessed one of the resources developed by the Ma’ayan Lab. The opacity of the blue color represents the number of users from each country.
As of November 2025, we published over 200 peer-reviewed publications co-authored with over 1,000 collaborators. These publications appear in top journals such as Nature, Science, Cell, PNAS, Nature Medicine, and Nature Cell Biology. These publications are the outcome of establishing positive working relationships with many world-class investigators. Many of our publications describe the original software that we have developed. The original data analysis and visualization software tools that we have developed are making a substantial impact on the biomedical research community for hypotheses generation and data analysis around the world.