
February 23, 2023
Monocytes re-enter the bone marrow during fasting and alter the host response to infection
Both overconsumption of food as well as fasting profoundly alter our immune system in opposite directions, with fasting being attributed anti-inflammatory effects. Recent studies had shown that fasting leads to redistribution of mature leukocytes throughout the body, raising more question about how dietary cues influence leukocyte production and trafficking. It also leads to the question what impact re-feeding after fasting has on the circulating leukocyte pool. In this study out in Immunity, led by @JanssenHenrike and @FlorianKahles, we focused on the mechanism and effects of fasting and re-feeding on monocyte dynamics and homeostasis.
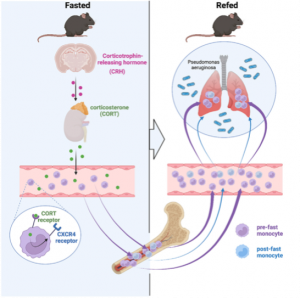
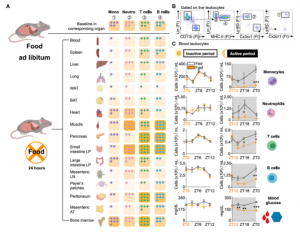
We first wanted to assess how a prolonged 24h fast would change the distribution of leukocytes throughout the body. Notably, every tissue except the BM had a reduced number of monocytes compared with the corresponding tissue of non-fasted animals. We performed a time-course analysis, asking whether the fasting effect on monocytes depended on the time of day. We found that food restriction during the active period (ZT12–ZT24/0) but not during the inactive period (ZT0–ZT12), rapidly reduced blood-glucose concentration, and circulating monocytes, suggesting that meals early after waking prevent drops in leukocyte numbers.
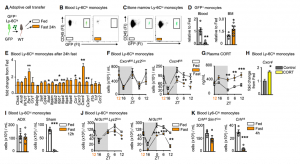
A drop of circulating leukocytes could be the result of newly produced monocytes being trapped in the bone marrow, increased cell death or remobilization of monocytes into other tissues, such as the bone marrow, where we had detected an increase of monocyte numbers. Considering that monocytes reduced dramatically from the circulation within a 4h hour fast, we hypothesized that monocytes were actively returning to the bone marrow, which was confirmed via adoptive transfer. The reverse mobilization was dependent on the upregulation of CXCR4, a leukocyte receptor essential of circadian migration. Fasting, as a form of stress, quickly led to the activation of the hypothalamic-pituitary-adrenal (HPA) axis, resulting in high plasma corticosterone (CORT) levels. CORT then bound to its receptor on monocytes, resulting in the upregulation of CXCR4 and homing of monocytes to the bone marrow.
Fasting ultimately leads to starvation if food is not reintroduced. To study the impact that re-feeding would have on the circulating monocyte pool, we reintroduced food after a 24h fast and assessed monocyte counts 4h later. Surprisingly, monocytes returned to the blood in much higher numbers, resulting in a surge of monocytes, presumably partially consisting of monocytes that had survived the fast in the bone marrow.
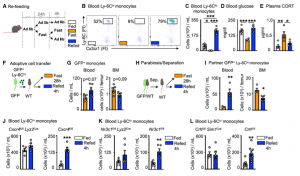
To further explore if in fact the surge of monocytes upon re-feeding consisted of older (produced before the fast) and younger (produced during the fast) cells, we labelled leukocytes with EdU before and with BrdU during the fast. This timed labelling would result in a pool of monocytes only positive for EdU and therefore produced prior to the fast (old) and a pool of monocytes only positive for BrdU, therefore produced during the fast (young). Upon re-feeding we saw that the surge of monocytes coming back into circulation consisted predominantly of older monocytes.

We sought to investigate if this pool was qualitatively different from ad libitum-fed conditions. We started by performing bulk RNA-seq of sorted blood monocytes retrieved after fasting/re-feeding and feeding ad libitum. As expected, differences were small. We used single cell RNAseq for further in depths analysis, and through pseudotime analysis confirmed that resurging monocytes upon re-feeding were older than under ad lib conditions.
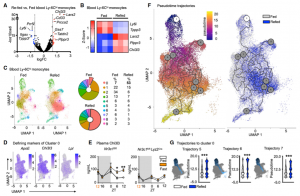
Having demonstrated that monocytes released from the BM upon re-feeding were chronologically older and transcriptionally different, as well as larger in numbers, we wondered if the older pool would dominate a site of acute inflammation. When mice were infected 4h after re-feeding in a model of pneumonia, higher numbers of monocytes were recruited into the parenchyma of the lung and ultimately resulting in higher mortality compared to mice that had food available ad lib.
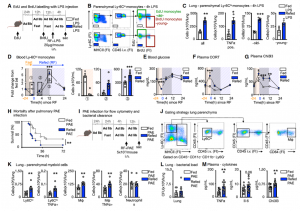
Our data reveal that prolonged fasting and re-feeding has significant impact on the circulating monocytic pool not only in respective to number but also in relation to disease outcome. Although the marrow is a safe haven for monocytes during nutrient scarcity, re-feeding prompted mobilization culminating in monocytosis of chronologically older and transcriptionally distinct monocytes. These shifts altered response to infection. Our study shows that diet—in particular, a diet’s temporal dynamic balance—modulates monocyte lifespan with consequences for adaptation to external stressors.
Read our complete paper with all the findings here.
June 2, 2022
Brain motor and fear circuits regulate leukocytes during acute stress
Acute stress is a physiological reaction to prepare the body for fight-or-flight situations. The emotion of stress is generated in the brain, where external and internal inputs are integrated and evaluated. Stress affects multiple organs systems, including the immune system. How the brain orchestrates the immune response to stress is poorly understood. Our laboratory has recently combined neuroscience, cardiovascular, and immunological techniques to investigate how distinct neuron clusters in the brain control neuro-immune pathways and peripheral immune cell distribution and function. Our data suggest that brain motor circuits induce rapid neutrophil mobilization from the bone marrow anticipate injury and associated infection. The paraventricular hypothalamus, conversely, controls a mass-migration of peripheral monocytes and lymphocytes to the bone marrow, which protects against the acquisition of autoimmunity, but impairs immunity to SARS-CoV-2 and influenza infection. The results of this study led by first author Dr. Wolfram Poller are published in Nature and we are excited to see how they open new avenues in brain-immune research related to cardiovascular disease and beyond.
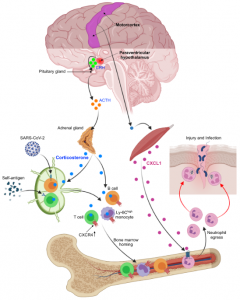
How the brain controls the immune system during acute stress has previously been surmised by manipulating intermediate conduits, but there has been little focus on processes linking brain networks with leukocyte dynamics.
We first characterized the effects of different psychosocial stressors and stress durations on blood leukocyte numbers. We observed massive counter-directional leukocyte shifts within hours of stress: whereas lymphocyte and monocyte numbers diminished gradually and returned to baseline during stress recovery, neutrophil numbers rose quickly and sharply returned to baseline. Remarkably, four hours of restraint stress reduced the circulating B cell, T cell, and Ly-6Chigh monocyte numbers by approximately 90%.
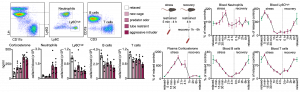
The effects of a single 4h restraint stress episode on lymphocyte numbers in the crucial secondary lymphoid organs lymph nodes and spleen were very long lasting. Even 40h of recovery was insufficient to replenish the secondary lymphoid organs to pre-stress numbers.
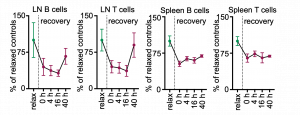
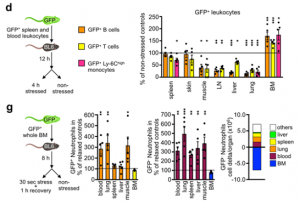
Using GFP cell tracking experiments, we showed that stress diverts lymphocytes and monocytes out of blood and peripheral organs and sequesters them in the bone marrow, while at the same time, neutrophils are strongly mobilized out of the bone marrow.
We next sought to elucidate the pathways via which the brain controls peripheral leukocyte dynamics. We started by testing the hypothalamic-pituitary-adrenal axis (HPA) and the sympathetic nervous system (SNS). Adrenalectomy, which ablated corticosterone release in stressed mice, abrogated the decrease in lymphocytes and monocytes in the blood whereas injecting corticosterone to control animals reduced cell numbers, indicating that the HPA was modulating these populations.
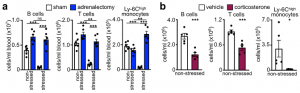
To determine whether the HPA acted directly on leukocytes via the glucocorticoid receptor (GC) (nuclear receptor subfamily 3, group C, member 1 (NR3C1)), we generated mice lacking NR3C1 in specific leukocytes. We found that the effects were leukocyte intrinsic.

The remaining minor decrease of B cells, T cells, and monocytes in the respective GR-KO lines during stress is likely attributed to the suboptimal efficacy of the Cre mouse lines used to delete the GR. We analyzed the efficiency of the Cre lines by crossing them to EYFP reporter mouse lines and found that CD19Cre activates EYFP expression in 88.1% of the B cells, CD4Cre in 91.6% of the CD4+ T cells and 93.8% of the CD8+ T cells, and LysMCre in only 38.0% of the Ly-6Chigh monocytes. These data indicate that around 11.9% of the B cells, 8.4% of CD4+ T cells, 6.2% CD8+ T cells, and 62% of the Ly-6Chigh monocytes still express the GR and therefore still disappear during stress.

Because CXCR4 is a bone marrow retention and homing factor regulated by glucocorticoids, we wondered whether lymphocytes and monocytes used this chemokine receptor. Indeed, B/T cells and monocytes augmented Cxcr4 expression during stress, and CXCR4 inhibition blocked the cells’ transit to the bone marrow. These data suggest that the HPA controls lymphocytes’ and monocytes’ stress-induced migration from peripheral tissues and blood to the bone marrow via leukocyte-intrinsic corticosterone-mediated augmentation of CXCR4.
To understand how the brain controls HPA-mediated leukocyte migration into the bone marrow during acute stress, we turned our attention to the paraventricular hypothalamus (PVH), which is active during acute stress, as indicated by the neuronal activation marker cFos.
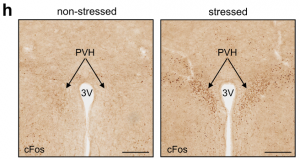
First, we chemogenetically stimulated CRH neurons in the PVH of CRHCre. Compared to controls, CRH neuron activation in this brain region increased peripheral corticosterone and strongly decreased lymphocytes and monocytes in the blood. Second, we performed the converse experiment by injecting an AAV encoding Cre-dependent expression of Caspase3 into the PVH of CRHCre mice. When stressed, mice lacking CRH neurons showed an abrogated corticosterone response and no more decrease in B cells, T cells, or monocytes. Third, we deleted the CRH gene from hypothalamic neurons by crossing CRHfl/fl mice with Sim1Cre mice and detected neither elevated corticosterone levels nor reduced B cells, T cells, or monocytes in the blood during stress, highlighting that the neuropeptide CRH rather than CRH neurons’ projections to downstream sites, induce leukocyte shifts. The PVH was not required for inducing neutrophilia; on the contrary, the PVH strongly blunted neutrophilia. Thus, the PVH/CRH axis is required for the reduction and bone marrow homing of circulating B cells, T cells, and monocytes during acute stress.
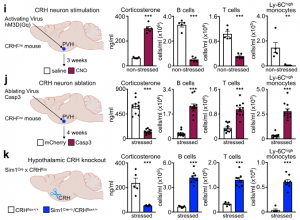
We next sought to determine which of the brain’s output systems was responsible for stress-induced neutrophilia. We hypothesized that the SNS was the likely culprit as it plays a crucial role in leukocyte mobilization. However, a series of gain- and loss-of-function experiments, including chemical SNS ablation, pharmacological blocking and genetic deletion of adrenoceptors, high dose noradrenaline injections, adrenalectomy, IL-6 knockout mice, and optogenetic stimulating of catecholaminergic neurons in the rostroventrolateral medulla (RVLM), all together indicate that acute restraint stress-induced neutrophilia was SNS-independent.
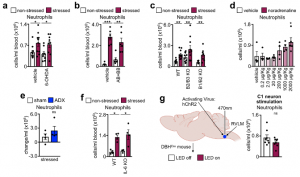
The finding that acute stress-induced neutrophilia was SNS-independent necessitated a different strategy. We measured a series of possible neutrophil modulators in the blood after restraint. Among them, two were increased, one of which was IL-16, which was present at low levels. The other was CXCL1, which is known to mobilize neutrophils, and which was abundantly present. The CXCL1 increase in the serum temporally correlated with blood neutrophilia, whereas an inhibitor of its receptor CXCR2 abrogated neutrophilia.
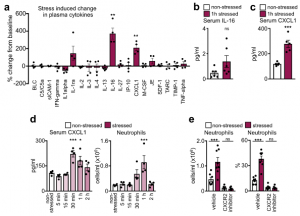
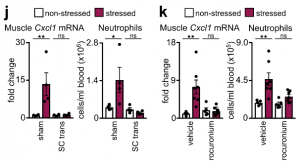
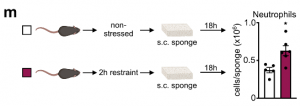
These data led us to identify the source of CXCL1 during restraint stress. Among the tissues, skeletal muscle showed the most augmented Cxcl1 expression. Cxcl1 mRNA increase in the muscle was very rapid, and closely followed by an increase in muscle CXCL1 protein. However, muscle Cxcl1 mRNA, serum CXCL1 protein, and blood neutrophils did not increase in response to voluntary running, suggesting that very strong muscle activation, as confirmed by EMG telemetry, was required. Circulating markers of muscle damage were not elevated after restraint stress suggesting that no major muscle injury had occurred.
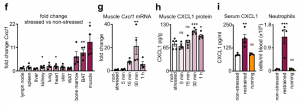
Investigating the mechanism of stress-induced muscle CXCL1 production, we found that spinal cord transection eliminated CXCL1 in the paralyzed skeletal muscle and prevented blood neutrophilia. Wondering whether motor innervation of the muscle itself was responsible for CXCL1 augmentation, we targeted the neuromuscular junction as the most downstream site using low doses of the non-depolarizing muscle relaxant rocuronium. At the dose used, rocuronium induced general muscle weakness and prevented excessive movement but did not interfere with normal respiration. This intervention blunted the stress-induced increase in muscle Cxcl1 and blood neutrophils, suggesting that central motor circuits control stress-induced neutrophilia.
To test the role of central motor circuits directly, we implemented optogenetics. Stimulating forelimb-related motor areas in the right motor cortex of Thy1-ChR2YFP mice, amplified Cxcl1 mRNA in the left but not the right biceps. The Cxcl1 increase was small, which is expected given the low strength of muscle movement induced by optogenetic stimulation under anesthesia. To induce stronger muscle movement, we optogenetically targeted motor centers in the medulla, where many motor circuits converge, leading to widespread skeletal muscle movement and strongly raising Cxcl1 mRNA in various skeletal muscles. Correspondingly, we found elevated CXCL1 in serum, along with blood neutrophilia.

Having demonstrated that stimulation of central motor structures induces CXCL1 and neutrophilia, we wondered whether inhibiting these structures during restraint stress prevents neutrophilia. First, bilaterally ablating the motor cortex slightly impaired muscle movement and blunted restraint stress-induced neutrophilia. Second, chemogenetically inhibiting glutamatergic neurons in the rostral medulla inhibited movement and prevented restraint-induced neutrophilia. By combining optogenetics with muscle relaxation we found that optogenetic medulla stimulation in the presence of a peripheral muscle relaxant no longer induced neutrophilia. Thus, neutrophilia arises as a consequence of central motor circuit-controlled excessive muscle activity.

Given such widespread and sweeping relocations of leukocyte during acute stress, we next inquired how leukocyte tissue distribution and function influence disease. We started with bulk RNAseq on blood neutrophils sorted under various stress conditions. Principal component analysis revealed distinct clustering among the groups with strongest expression differences in important neutrophil function-related genes appearing after 1 h of stress, with diminishing differences between non-stressed and recovering animals. Hierarchical clustering showed transcripts that changed in both directions after 1 h of stress and that either returned to pre-stress levels or remained altered even after 3 h of recovery. Crucial immunological pathways were enriched after stress.
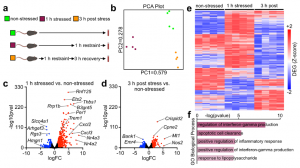
To examine stress-induced changes among neutrophil subsets, we performed single-cell RNA sequencing. We noted at least four neutrophil clusters, some of which changed in response to stress. Among them, a cluster defined by neutrophil degranulation, activation, and aggregation, expanded in response to stress. Some of these clusters resembled those described in a recent single-cell analysis of neutrophil heterogeneity in the blood. Moreover, we noted that stress accelerates neutrophil maturation, according to a neutrophil maturation score. Comparing key neutrophil mediators further bolstered the idea that acute stress elevates neutrophils’ capacity to modulate inflammation.

In line with these sequencing data, we observed that neutrophils indeed readily accumulated at injury sites, which is in keeping with previous findings.
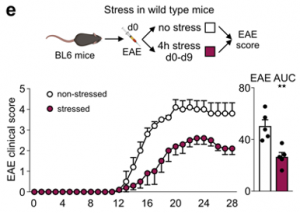
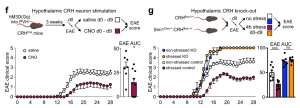
To assess potential implications of stress-induced leukocyte redistribution from the lymph nodes to the bone marrow, we utilized models of autoimmunity and viral infection. Although the strong reduction in the number of adaptive immune cells in lymph nodes during acute stress might appear to be detrimental, restricting the acquisition of adaptive immunity may be beneficial under certain conditions. We began testing these ideas by subjecting mice to experimental autoimmune encephalomyelitis (EAE). Consistent with earlier data, we found fewer B and T cells in the lymph nodes of EAE mice subjected to stress but more B and T cells in the bone marrow. We enumerated leukocytes in the spinal cord and found considerably less inflammation in mice that were stressed during sensitization.

Consequently, mice subjected to stress had lower EAE clinical scores and were protected from disease initiation and progression.
To link stress’s influence on the acquisition of autoimmunity with relevant brain regions, we applied chemogenetics. We stimulated CRH neurons in the PVH during the induction phase of EAE and found less severe disease initiation compared to controls. Conversely, we tested the effect of stress on EAE initiation in mice that genetically lacked CRH in the hypothalamus and found that the protective effect of stress on acquisition of EAE was abrogated. Compared to controls, mice lacking hypothalamic CRH were highly susceptible to EAE even when stressed. These data show that stress activates specific brain regions that prevent the acquisition of autoimmunity.
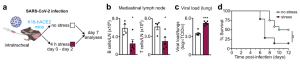
To test how lymphocyte redistribution from the lymph nodes to the bone marrow affects immunity to viral infections, we infected K18-hACE2 mice with SARS-CoV-2 and subjected them to episodes of acute restraint stress. We observed fewer B cells and T cells in the lung-draining mediastinal lymph nodes on day 7. Compared to controls, the stressed mice had higher viral titers and died at higher rates from SARS-CoV-2 infection.
We observed similar effects in stressed mice subjected to influenza A virus (IAV) infection and dug deeper into the mechanism using this model. First, we found that selective stimulation of PVH CRH neurons recapitulated the long-lasting effect of stress on lymph node cellularity, IAV-specific antibody titers, and pulmonary viral loads, whereas selective ablation of PVH CRH neurons prevented the detrimental effect of stress on these parameters. Second, we explored the relevant immune cells and receptors and observed no effect of stress on viral loads in Rag1, Tcr𝛼 KO and 𝜇MT mice. We infected CD19Cre+/-:GRflox+/+ and CD4Cre+/-:GRflox+/+ mice and saw no stress-induced changes in the number of the respective cell type in the mediastinal lymph nodes; mice lacking GR on B cells exhibited no stress-induced decrease in the IAV specific antibody titers in the BAL and neither GR KO line showed stress-induced increases in pulmonary viral loads.
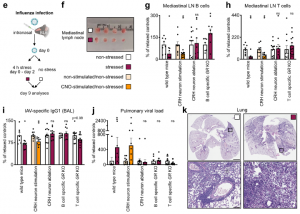
These data reveal a pathway linking stress-induced PVH CRH neuron activity with corticosterone acting directly on lymphocytes, thus impairing sensitization to viral infection, and aggravating disease.
Our study reveals how specific neuron clusters located in diverse regions of the brain relevant to fear and the fight or flight response cause massive changes in leukocyte distribution and function. Future studies are required to deepen our understanding of these neuroimmune pathways.
Read our complete paper with all the findings here: https://www.nature.com/articles/s41586-022-04890-z
July 16, 2021
Astrocytic interleukin-3 programs microglia and limits Alzheimer’s disease
Despite being responsible for the majority of dementia cases, which inflict approximately 50 million people globally, Alzheimer’s Disease remains poorly understood in its causes and its progression. Our laboratory recently investigated immune factors involved in Alzheimer’s Disease and discovered an unexpected role for IL-3 signaling in amelioration of Aβ burden and cognitive decline. Our data suggest that this phenomenon is conserved in mice and humans and, thus, open the door for potential immunotherapies targeting this axis. Our results are out now in Nature and we are excited to see what avenues of Alzheimer’s Disease research they open up. Thanks to the wonderful collaboration with the Tanzi Lab and all others involved. Check out the excellent Nature News and Views highlighting our study.
This work was headed by first author Dr. Cameron McAlpine, who has recently started his own lab here at the Icahn School of Medicine and is actively recruiting post-docs. Please apply to those interested. See below for greater explanation by @cam_phd into our findings:
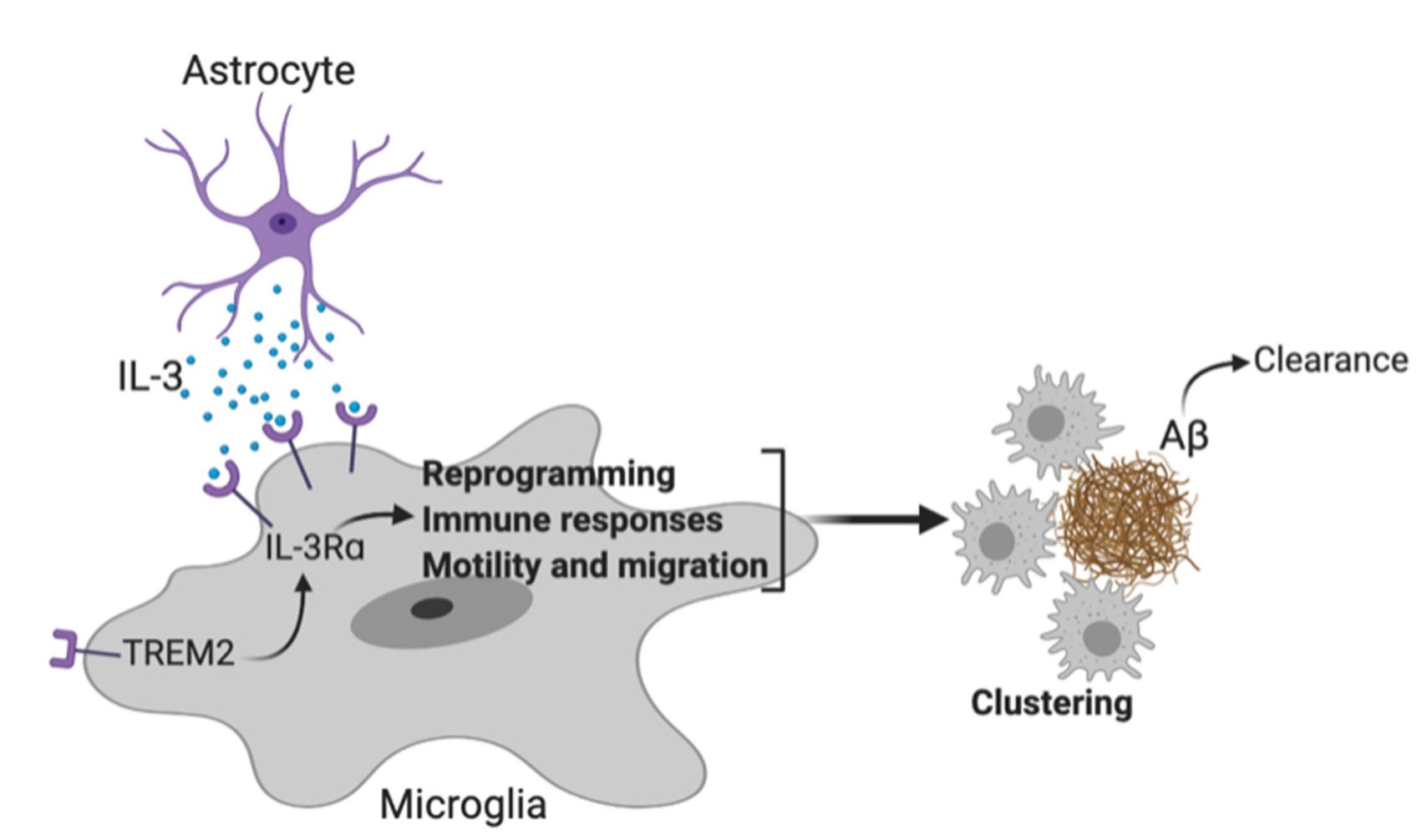
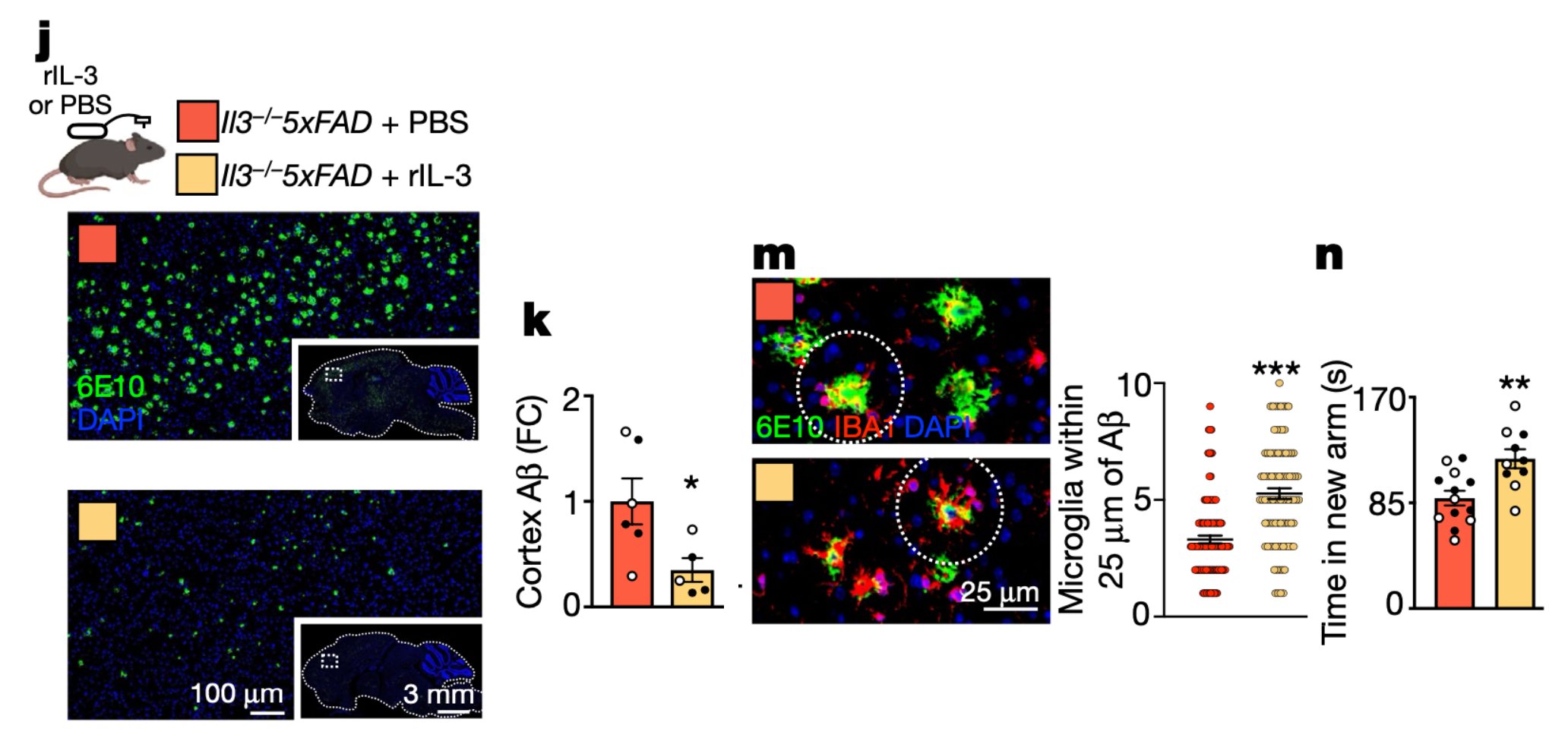
IL-3 is a multifunctional cytokine and an immune regulator. Despite some hints, the role of IL-3 in Alzheimer’s disease (AD) was unknown. We found that IL-3 deletion worsens AD pathology and cognitive decline in 5xFAD mice, a murine model of AD.
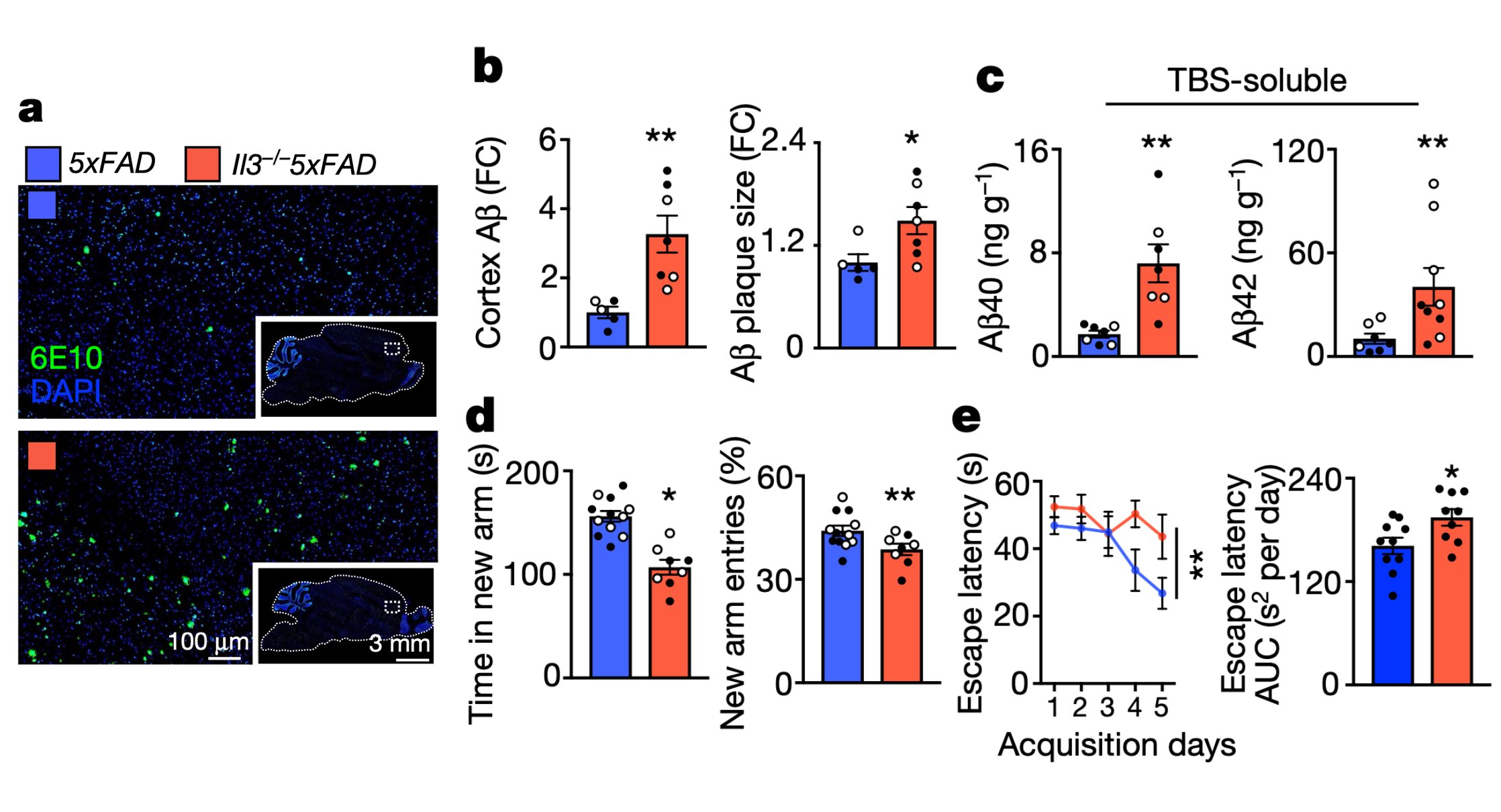
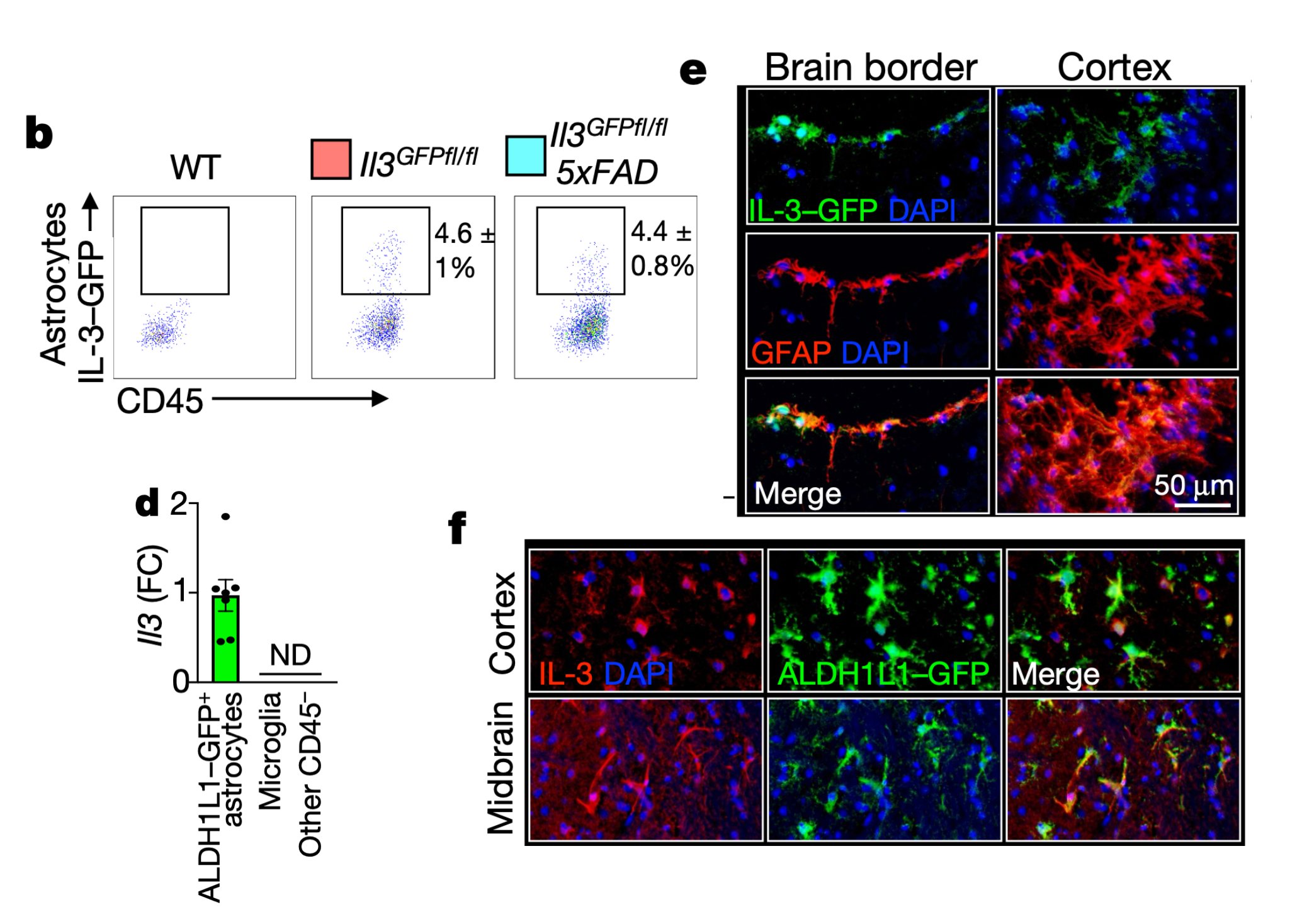
We then generated IL-3 reporter/flox mice using a CRISPR-Cas9-based editing strategy and found that a subset of astrocytes, but not other brain cells, generate IL-3.
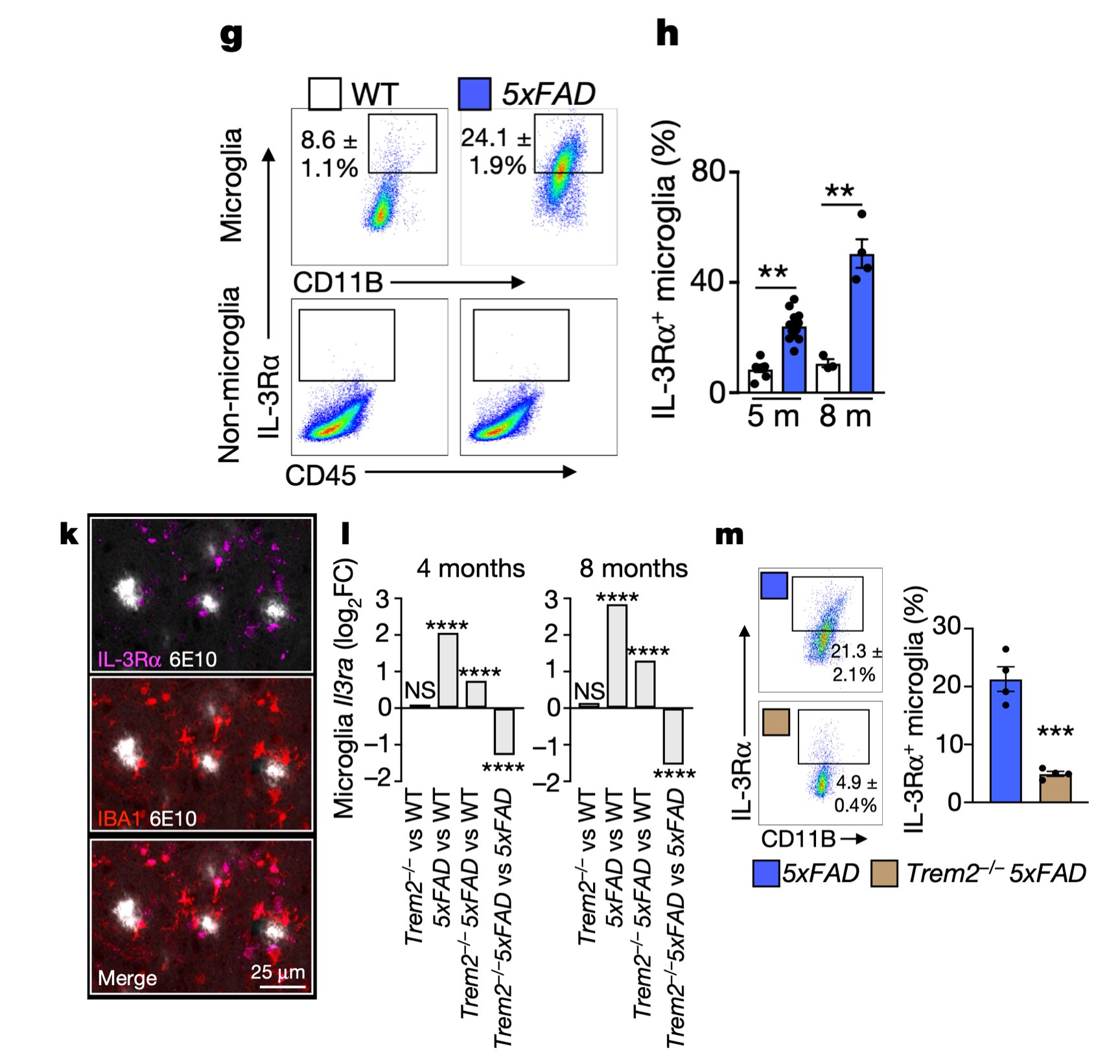
Interestingly, in AD, microglia become responsive to this astrocyte-sourced IL-3 by augmenting expression of IL-3’s receptor, IL-3Rɑ, via TREM2 signaling
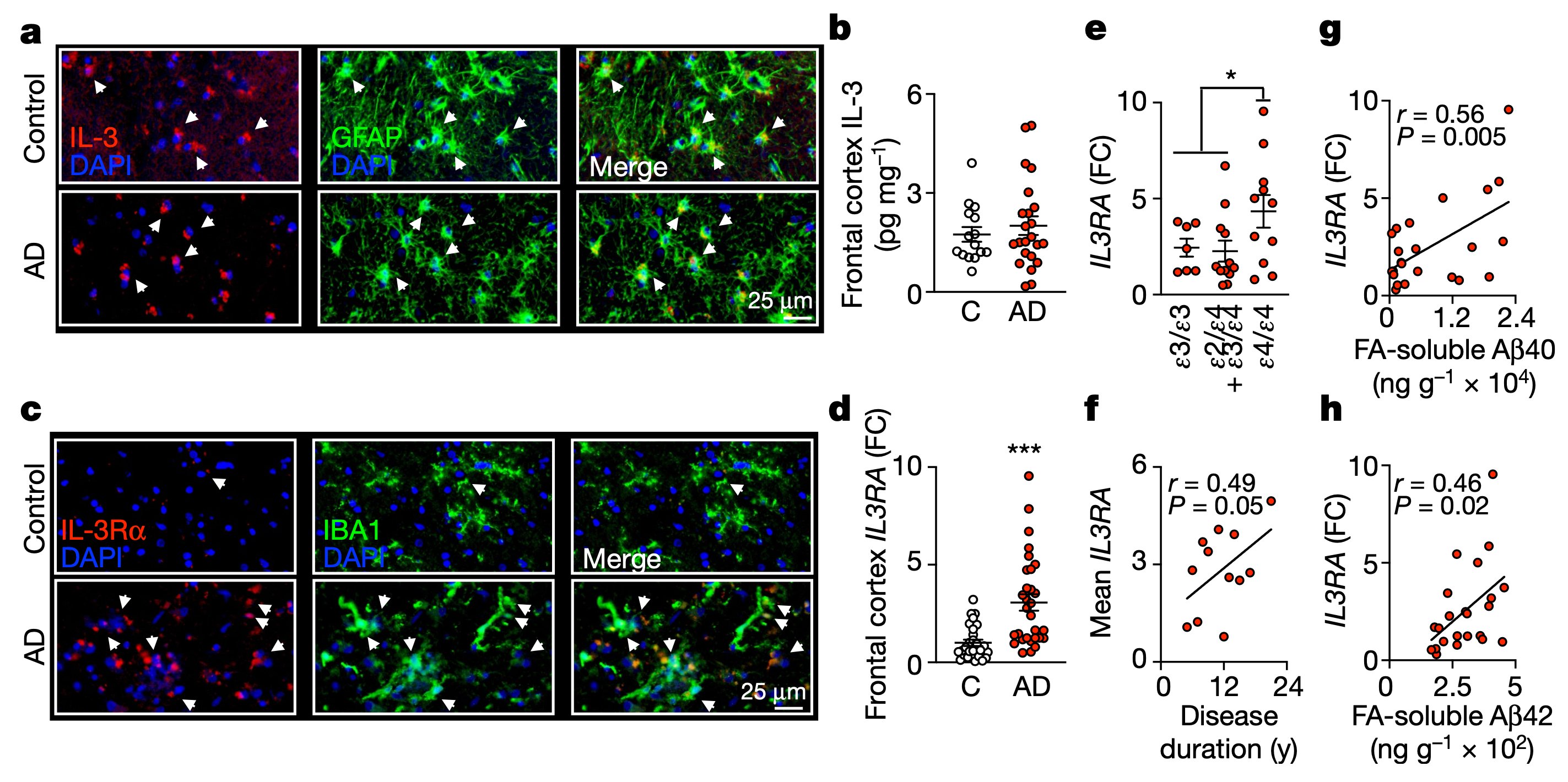
This Dynamic IL-3 signaling is not only relevant in mice but also occurs in humans with AD. In fact, IL-3RA expression correlates with APOE genotypes and Aβ load in the brain of AD patients.
Finally, to explore therapeutic potential, chronic infusion of recombinant IL-3 into the murine brain ameliorated AD.
Read our complete findings here: https://www.nature.com/articles/s41586-021-03734-6
And check out extended media coverage here: https://www.eurekalert.org/pub_releases/2021-07/mgh-ris071221.php