
We have developed the following suite of bioinformatics software tools and web applications for analyzing and acccessing CPTAC proteogenomic data:
Database of Genes Related to Platinum Resistance
ProTrack
Interactive heatmap visualizations for individual CPTAC cohorts
ProTrack applications can be found at: http://protrack.cptac-data-view.org/
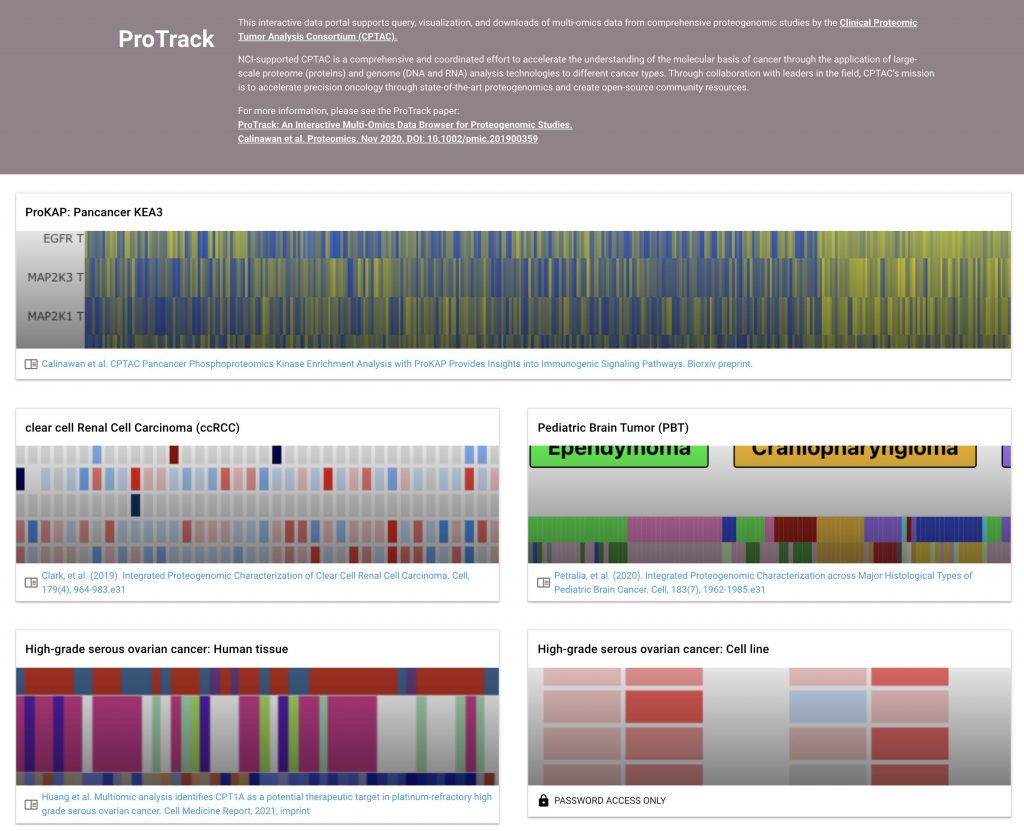
The Clinical Proteomic Tumor Analysis Consortium (CPTAC) initiative has generated extensive multi-omics data resources of deep proteogenomic profiles for multiple cancer types. To enable the broader community of biological and medical researchers to intuitively query, explore, and download data and analysis results from various CPTAC projects, we present the user-friendly web application called “ProTrack”
iProFun
iProFun provides integrative analysis results for identifying DNA-level alterations perturbing functional molecular traits.
iProFun results for published CPTAC cohorts are available at http://www.cptac-iprofun.org/ .
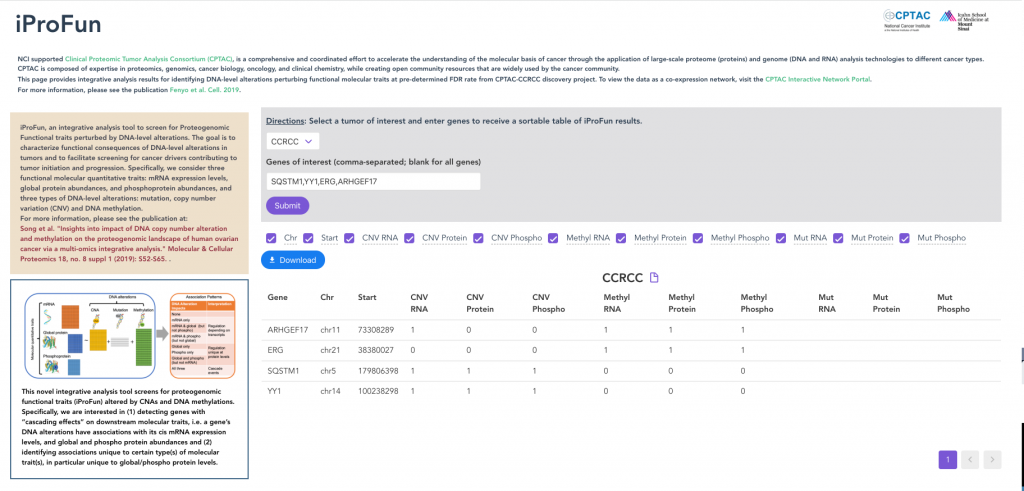
We consider three functional molecular quantitative traits: mRNA expression levels, global protein abundances, and phosphoprotein abundances. We aim to identify those genes whose CNAs and/or DNA methylations have cis-associations with either some or all three types of molecular traits.
We applied iProFun to multiple CPTAC cancer types. Users can enter genes and view which have CNA and/or DNA methylation has cis-associations with its mRNA expression, global protein, and phosphoprotein abundances.
The R package and source code are also available on Github: https://github.com/WangLab-MSSM/iProFun
ProKAP
Interactive visualizations for CPTAC pan-cancer kinase enrichment analysis results.
Kinase activity scores are available at: http://pancan-kea3.cptac-data-view.org/
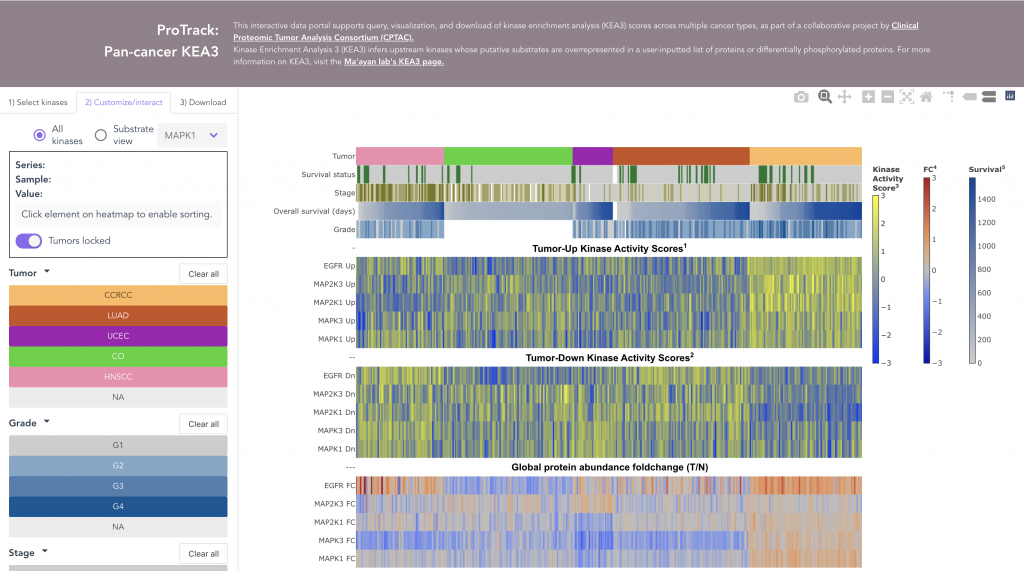
We performed Kinase Enrichment Analysis (KEA) using a CPTAC phosphoproteomics dataset to identify putative differences in kinase state between tumor and normal tissues within and across five types of cancer.
The ProTrack Kinase Activity Portal (ProKAP) is an interactive web application for querying, visualizing, and downloading the derived pan-cancer kinase activity scores together with the corresponding sample metadata, and protein and phosphoprotein expression profiles.
Ref (Biorxiv preprint)
CPTAC Pancancer Phosphoproteomics Kinase Enrichment Analysis with ProKAP Provides Insights into Immunogenic Signaling Pathways. Calinawan et al. Nov 2021. Biorxiv preprint.
BayesDeBulk
BayesDeBulk can be utilized to perform bulk deconvolution beyond transcriptomic data, based on other data types such as proteomic profiles or the integration of both transcriptomic and proteomic profiles.
This webtool performs tumor deconvolution on an input expression table and/or protein abundance file with cell types from pre-populated, published cell signatures.
The website can be found at: https://calina01.u.hpc.mssm.edu/bayesdebulk/
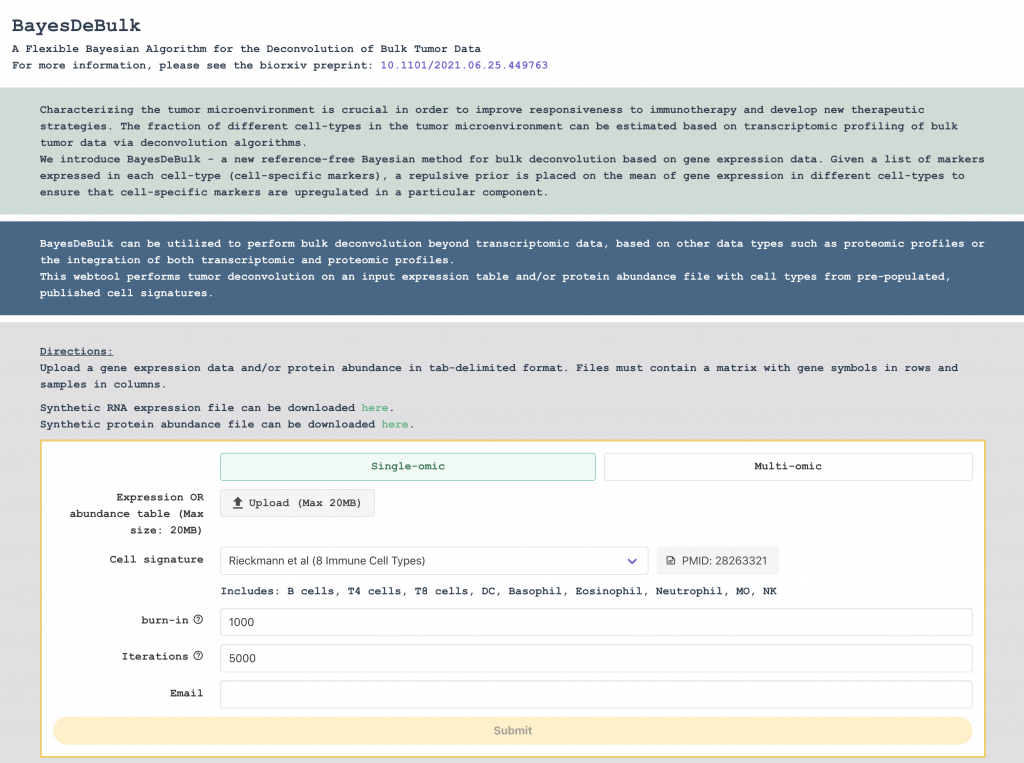
BayesDeBulk – a new reference-free Bayesian method for bulk deconvolution based on gene expression data. Given a list of markers expressed in each cell-type (cell-specific markers), a repulsive prior is placed on the mean of gene expression in different cell-types to ensure that cell-specific markers are upregulated in a particular component. Contrary to existing reference-free methods, the labeling of different components is decided a priori through a repulsive prior.
BayesDeBulk can be utilized to perform bulk deconvolution beyond transcriptomic data, based on other data types such as proteomic profiles or the integration of both transcriptomic and proteomic profiles.
Ref (Biorxiv preprint)
BayesDeBulk: A Flexible Bayesian Algorithm for the Deconvolution of Bulk Tumor Data. Petralia et. al. June 2021. Biorxiv preprint.
Database of Genes Related to Platinum Resistance
As a resource to the cancer research community, we provide a comprehensive overview accompanied by a manually curated database of the >900 genes/proteins that have been associated with platinum resistance over the last 30 years of literature.
The browsable database can be found at: http://ptrc-ddr.cptac-data-view.org/#/
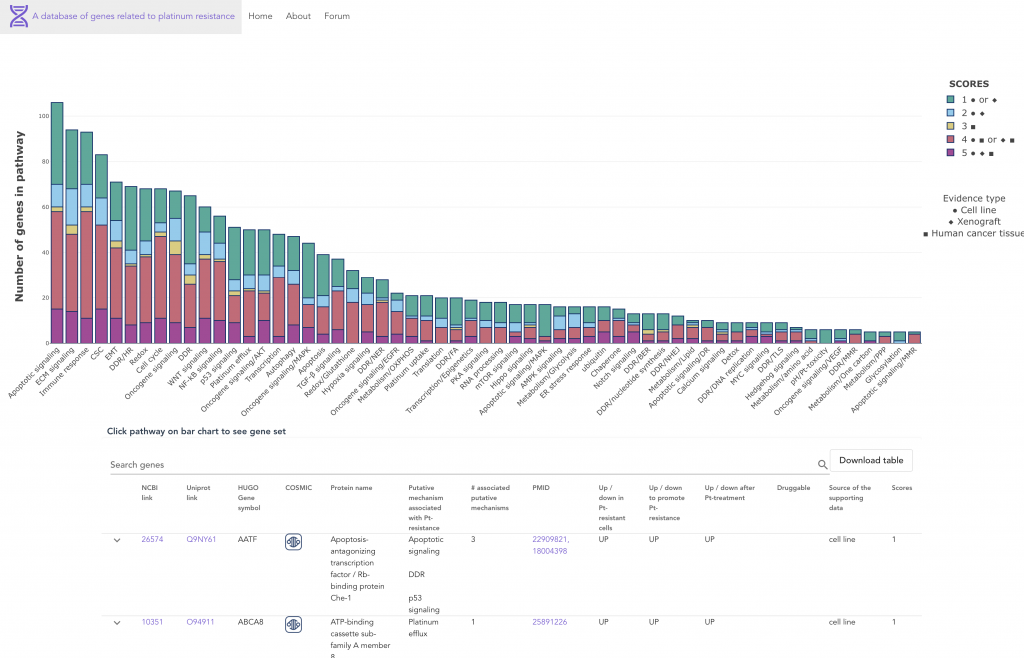
Extensive research has been conducted to understand and overcome platinum resistance, and mechanisms of resistance can be categorized into several broad biological processes, including (1) regulation of drug entry, exit, accumulation, sequestration, and detoxification, (2) enhanced repair and tolerance of platinum-induced DNA damage, (3) alterations in cell survival pathways, (4) alterations in pleiotropic processes and pathways, and (5) changes in the tumor microenvironment.
As a resource to the cancer research community, we provide a comprehensive overview accompanied by a manually curated database of the >900 genes/proteins that have been associated with platinum resistance over the last 30 years of literature.
DreamAI
An ensemble based imputation algorithm for labelled proteomics data resulted from the NCI-CPTAC DREAM Proteogenomics Challenge (2016) and post Challenge community effort.
The DreamAI R package can be downloaded at: https://github.com/WangLab-MSSM/DreamAI
In DreamAI, an ensemble imputation matrix is obtained from averaging results of six imputation algorithms: top 3 teams in challenge (spectroFM: Team DMIS_PTG; RegImpute: Team Jeremy Jacobsen; Birnn: Team BruinGo) and 3 baseline algorithms (KNN, missForest, ADMIN).
ProNetView
ProNetView-ccRCC (http://ccrcc.cptac-network-view.org/) is an interactive web-based network exploration portal for investigating phosphopeptide co-expression network inferred based on the CPTAC clear cell renal cell carcinoma (ccRCC) phosphoproteomics data is introduced.
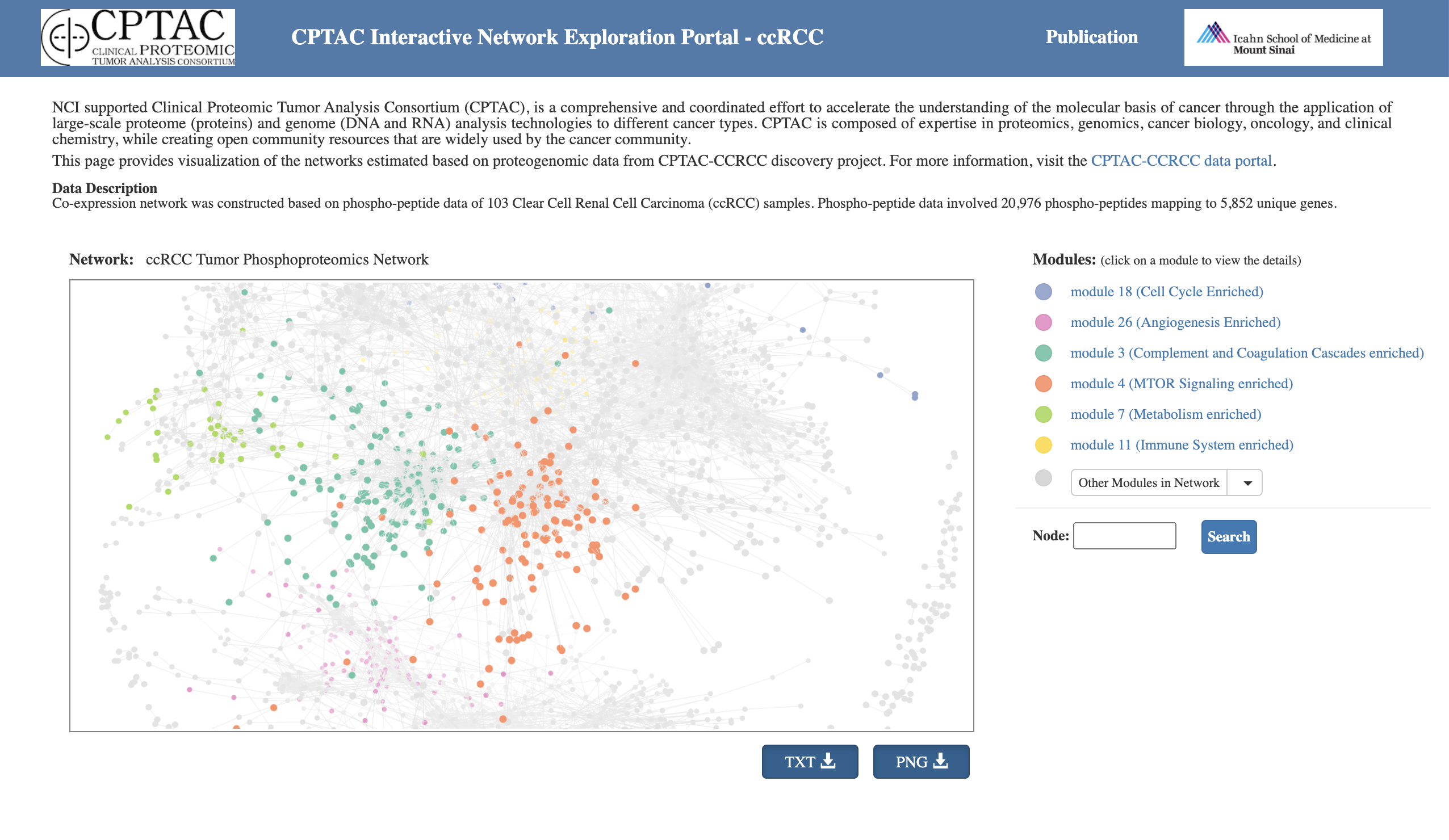
ProNetView-ccRCC enables quick, user-intuitive visual interactions with the ccRCC tumor phosphoprotein co-expression network comprised of 3614 genes, as well as 30 functional pathway-enriched network modules. Users can interact with the network portal and can conveniently query for association between abundance of each phosphopeptide in the network and clinical variables such as tumor grade.
iJRFNet
Integrative Joint Random Forest (iJRF) characterizes the regulatory system between miRNAs and mRNAs using a network model. iJRF is designed to work under the high-dimension low-sample-size regime, and can borrow information across different treatment conditions to achieve more accurate network inference. It also effectively takes into account prior information of miRNA–mRNA regulatory relationships from existing databases.
The R package can be downloaded from the CRAN network: https://cran.r-project.org/web/packages/JRF/index.html
The R package is also available on Github at: https://github.com/WangLab-MSSM/iJRFNet