
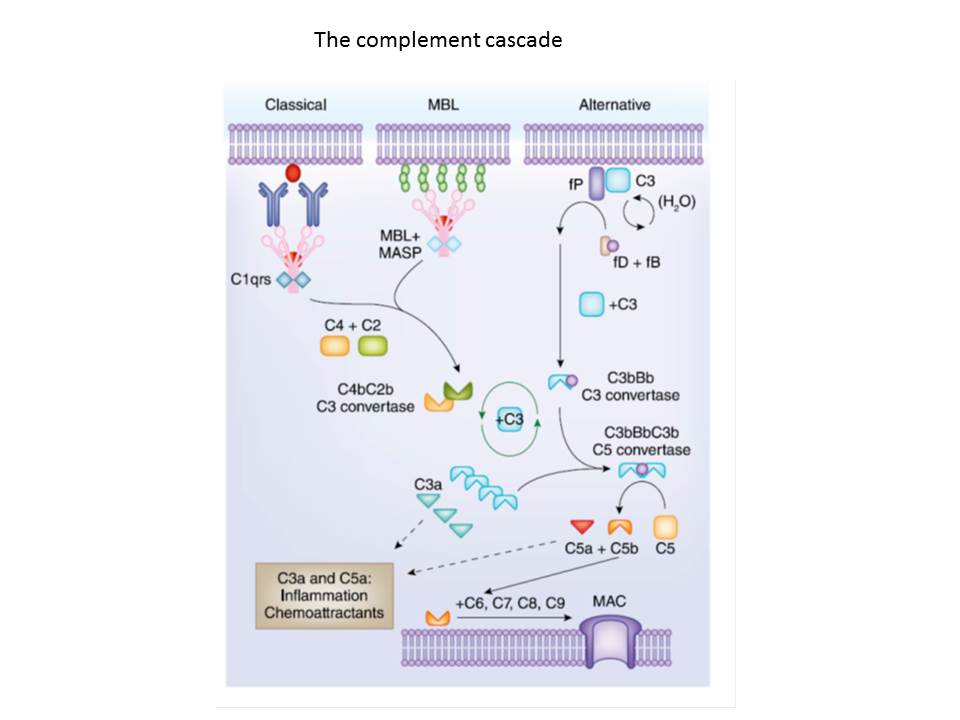
The basic science portion of the laboratory studies mechanisms of T cell alloreactivity and transplant tolerance with a focus on interactions between the complement system and T cell immunity. While significant efforts are being placed on developing methods to harness Foxp3+ T regulatory cells (Tregs) to prolong and possibly even prevent transplant rejection, optimization of approaches to reach these goals is required. Remarkably, our combined published and preliminary data demonstrate that signaling via murine and human CD4+ cell expressed C3a receptor (C3aR) and C5a receptor (C5aR) inhibit Treg induction, activation, and function whereas blockade markedly augments Treg induction, activation, function and stability. Our findings support the central hypothesis that C3a/C5a that are locally generated from immune cells function as physiological regulators of alloimmunity. The C3a and C5a bind (paracrine/autocrine) to C3aR and C5aR expressed on naïve T cells and thymic derived, natural Treg (nTreg), activating signaling cascades that prevent Foxp3 expression, together inducing Teff differentiation and expansion, at the same time inhibiting Treg induction, function and stability. This results in an expanded, pathogenic alloreactive T cell response that prevents prolonged graft survival and precludes tolerance induction. Our preliminary data support the hypothesis that blocking C3aR/C5aR signaling facilitates the generation of inducible Treg (iTreg), and enhances nTreg and iTreg function and stability. As the absence of C3aR/C5aR signaling simultaneously inhibits Teff differentiation/expansion, the overall effect is to limit pathogenic alloimmunity, prolong transplant survival, and according to our data facilitate tolerance induction. To decipher the connection of silenced C3aR/C5aR signaling with CD4+ Treg suppression of transplant rejection, our strategy will be to supplement in vitro studies with in vivo murine heart transplantation (that naturally results in rejection) and murine corneal transplantation (which naturally leads to tolerance). We hypothesize that performing such complementary experiments will provide molecular targets and methods that can be exploited to prolong transplant survival first in mice, then ultimately in humans. Additional studies are addressing the role of complement as a mediator of early posttransplant inflammation that activates Teff and inhibits Treg, and to assess interactions between complement and TLR signaling, that together prevent tolerance induction.
Regarding clinical and translational studies: Transplantation is the therapy of choice for patients with end stage kidney disease, but long-term allograft survival and function for recipients of deceased donor kidneys with graft half-lives of only ~10 years. Current thinking is that the poorer long-term survival of deceased donor kidneys is due in part to the short- and long-term consequences of ischemia reperfusion (IR) injury although the underlying mechanisms are incompletely understood. Epidemiological evidence strongly associates IR-induced delayed graft function (DGF) with an increased risk of early acute rejection. Evidence from animal models and from studies of human transplant recipients suggest that inflammation initiated by IR injury and DGF augments graft immunogenicity, thereby facilitating activation and amplification of pre-existing (memory), and de novo, graft-reactive, pathogenic T and B cell immunity, which together have negative long-term consequences for the allograft. The data derived from preclinical models strongly support the hypothesis that the immediate, posttransplant (IR-initiated), proinflammatory mediators that drive this process include complement, endogenous toll like receptor (TLR) ligands and innate cytokines, including tumor necrosis factor alpha (TNFa), which synergistically amplify graft injury through autocrine and paracrine ligations with receptors expressed on graft cells and on infiltrating immune cells. The hypothesis predicts that early posttransplant TNFa blockade will dampen the vicious cycle of allograft inflammation and as a consequence confer long-term protective effects on allograft function and survival. Through a multicenter clinical trial we are testing the hypothesis that in humans IR-induced TNFa production within the allograft at the time of transplantation plays a crucial role in amplifying local inflammation, enhancing activation, expansion and graft infiltration of donor-reactive T cells and B cells (the latter of which also produce donor-reactive antibodies), while simultaneously inhibiting regulatory T cell induction and function. Together with early induction of a fibrogenic molecular profile in the graft, the result is worsened graft function and poorer graft survival. Associated studies will define mechanisms of the intervention and identify novel biomarkers capable of assessing risk of injury and graft failure.