
One of the most fascinating aspects of cancer biology is the drastic change in cellular physiology that accompanies the acquisition of oncogenic mutations. Such extensive pathway rewiring, while necessary to accommodate unrestrained proliferation or metastatic behavior, might come at a cost for cancer cells, in the form of discrete genetic liabilities. Targeting these cancer-specific vulnerabilities might lead to system collapse, highly selective cancer cell death, and safer and more effective cancer therapies.
Conceptually, a molecular liability in cancer can be refined to a gene whose inactivation modifies the phenotype afforded by a cancer mutation (say, to convert a survival advantage into an apoptotic fate), but has otherwise little or no effect in wild-type cells throughout the body. Unfortunately, the lack of an adequate model system in which to identify such genetic modifiers on a systematic basis has hindered the discovery of rational targets in cancer.
Modeling Targeted Cancer Therapeutics in Zebrafish
To identify molecular liabilities associated with specific cancer genotypes, we take genetic and chemical-genetic approaches in the zebrafish system, complemented with validation studies in cultured human cancer cell lines. Like mice, zebrafish are vertebrates that faithfully recapitulate the core pathways of human oncogenesis. Decisively, however, zebrafish also offer the high-throughput capacity and genetic tractability of flies or worms. Another key advantage of the zebrafish model for identifying targets for systemic inhibition in humans is the system’s whole-body capacity, especially at the embryonic stage. Genetic or pharmacologic manipulation of large clutches of externally developing embryos not only enables target discovery, but also affords preclinical studies in which the therapeutic versus toxic effects of drugs can be analyzed with unparalleled spatial resolution in intact animals (Figs. 1,2). Once we identify a promising target, we can unravel its mechanism of action through morpholino-based epistasis analysis in live embryos, and readily assess the human relevance of our findings via RNAi and biochemical studies in cancer cell lines (Figs. 3,4).


 Figure 1. Target discovery via chemical genetics. p53 mutant zebrafish embryos are resistant to ionizing radiation (IR)-induced cell death (compare C and B), providing an animal model of treatment resistance in human tumors. Chk1 inhibition restores wild-type levels of IR-induced cell death in p53 mutants (compare F and C) but is otherwise non-toxic (note lack of acridine orange uptake in D). In contrast, Chk2 inhibition has no therapeutic effect in p53 mutants (compare I and C) at a dose that is already toxic (compare G and A).
Figure 1. Target discovery via chemical genetics. p53 mutant zebrafish embryos are resistant to ionizing radiation (IR)-induced cell death (compare C and B), providing an animal model of treatment resistance in human tumors. Chk1 inhibition restores wild-type levels of IR-induced cell death in p53 mutants (compare F and C) but is otherwise non-toxic (note lack of acridine orange uptake in D). In contrast, Chk2 inhibition has no therapeutic effect in p53 mutants (compare I and C) at a dose that is already toxic (compare G and A).
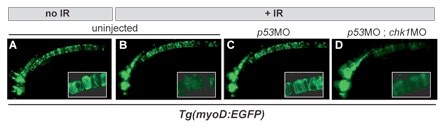
Figure 2. High-resolution in vivo imaging with cell/tissue specific transgenic reporter lines. Chk1 inactivation overcomes radioresistance caused by p53 loss in GFP-labeled notochord cells (compare C and D). Inset, high-mag of the notochord outlining individual cells. MO, morpholino (gene knockdown-inducing antisense oligonucleotide).
Taking this integrated approach, we previously discovered that vertebrate cells deficient in the p53 tumor suppressor gene are hyperdependent on the Chk1 protein kinase for survival after radiation-induced DNA damage (Sidi et al., Cell 133:864-877, 2008). Genetic or pharmacologic inhibition of Chk1 is sufficient to restore an apoptotic response to ionizing radiation (IR) in otherwise radioresistant p53 mutant zebrafish embryos or human cancer cells (Figs. 1-3), thereby overriding a major mechanism by which tumor cells evade radiation therapy (p53 is mutated in over 50% of human solid tumors). Surprisingly, we found that the mechanism by which Chk1 inhibition restores apoptosis in p53-deficient cells does not rely on reactivation of classical mitochondrial or death-receptor signaling downstream of malfunctional p53. Instead, Chk1-inhibited cells appear to trigger a fundamentally new form of apoptosis, designated “Chk1-suppressed” pathway, which involves the DNA damage-response kinases, ATM and ATR, and the highly conserved but poorly understood caspase-2 protease (Fig. 4).



Figure 3. Validation studies in human cancer cells. Immunoblotting, apoptotic assays (e.g. TUNEL), and RNAi are to assess the evolutionary and disease significance of the zebrafish data.


Figure 4. The vertebrate apoptotic response to DNA damage. Schematic highlighting the novel “Chk1-suppressed” pathway (red arrows), which operates independently of the classical mitochondrial and death-receptor pathways (middle and left, respectively). See Sidi et al. 2008 for details.
Chk1 as a Target: Breaking Down a Novel Apoptotic Pathway
The Chk1-suppressed (CS) pathway might define both a novel apoptotic mechanism and a uniquely broad strategy for the treatment of cancer. However, aside from its ATM/ATR-caspase-2 core backbone, we know virtually nothing of this pathway’s molecular and cellular circuitry (Fig. 4). Detailed knowledge of the CS pathway not only promises to illuminate a novel apoptotic device in our cells; it should also identify potential resistance mechanisms and predictive biomarkers in the pathway, both of which are mandatory for taking the pathway into the clinic. Thus a major focus in the lab is to identify novel CS pathway components. We are taking a systematic and unbiased approach involving a genome-wide RNAi screen in cell-culture and in vivo zebrafish genetics. In pilot screens, we have identified five new CS pathway members. Three of these genes are required for caspase-2 activation downstream of ATM and ATR, while the remaining two act downstream of caspase-2. The goal is now to functionally characterize these genes, examine their therapeutic implications, and pursue our large-scale screening efforts for additional players in the pathway.
Identification of Novel Cancer Targets through In Vivo Synthetic Lethality Screens
A second focus in the lab is to identify novel cancer liabilities by applying the genetic concept of synthetic lethality to cancer-relevant genotypes. As originally described in yeast, two genes are said to be synthetically lethal if mutation of either alone is viable but simultaneous mutation of both genes is lethal. This concept provides an attractive framework for identifying optimal targets for cancer therapy, because targeting a gene synthetically lethal to a cancer lesion should be deadly to cancer cells but harmless to all other cells in the body. The whole-animal and high-throughput capacities of the zebrafish embryo make it an ideal model system in which to identify synthetic lethal interactors of common molecular alterations in cancer, and we recently obtained proof of principle for the strategy (Fig.5). We are currently focusing on identifying synthetic lethal interactors of the PTEN tumor suppressor by examining the zebrafish orthologs of genes essential for the viability of PTEN mutant, but not PTEN wild-type, cultured cancer cell lines, in collaboration with Pr Alan Ashworth (ICR, UK).


Figure 5. Synthetic-lethal screening. A simple embryo viability assay, compatible with large-scale screening, suffices to identify a synthetic lethal interaction between FANCD2 and Chk1. See Chen, Kennedy, Sidi et al. 2009 for details.