




Small Cell Lung Cancer (SCLC)
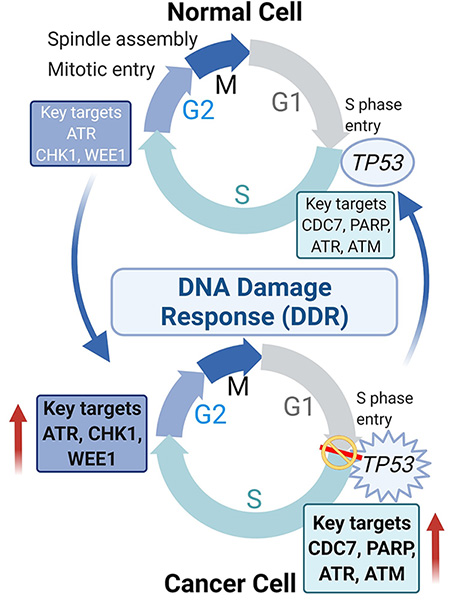
DNA damage response (DDR) as a transformative therapeutic vulnerability in SCLC
Our comprehensive preclinical studies demonstrated the effectiveness of targeting DNA damage response (DDR) proteins like CHK1 and WEE1 inhibition in SCLC which has supported clinical testing of these inhibitors in SCLC patients. Additional preclinical data explored potential combinations of CHK1 inhibitor and chemotherapy and has identified MYC as a predictive biomarker of response to CHK1 inhibition in SCLC (Sen et al, 2017a). We established AKT/mTOR signaling as a mechanism of innate and acquired resistance to WEE1 inhibition in SCLC.
Current projects in the lab include: (1) validating the effectiveness of additional DDR proteins like ATR, ATM and DNAPKCs as a tractable SCLC target; (2) identifying mechanisms of resistance to DDR inhibition and exploring potential combination treatments to over resistance; and (3) discovery of SCLC subsets defined by the transcriptional signature that predicts improved responses to DDR targeted drugs. We are currently working other institution and industry partners to design biomarker-driven DDR inhibitor trials for SCLC.
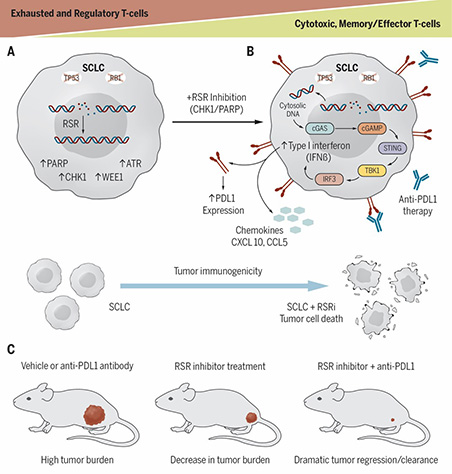
Modulators of replication stress and their role in immune evasion
Immune checkpoint blockade (ICB) like anti-PD-1 and anti-PD-L1, have been at the forefront of clinical practice for treating several solid tumors. Despite this success, most patients fail to respond to ICB therapy due to innate or acquired resistance. Previous reports have demonstrated that cancers with higher genomic instability and RS respond better to immunotherapy. However, there is limited understanding of the role of DDR in activating the immune system. Further, the clinical activity of ICB in patients with DDR altered tumors has not been well described. We provided the first evidence that targeting DDR proteins can activate the immune microenvironment in SCLC, and that activation happens through the innate immune cGAS/STING pathways which leads to remarkable synergy when combined with ICB. Collectively, the research highlighted the intimate crosstalk between the cancer cells and the tumor microenvironment, and raised several fundamental questions: What are the signaling pathways in the tumor microenvironment regulated by DDR pathways? What is the therapeutic utility of targeting DDR pathways in the treatment of cancer progression and immune evasion?
We are currently investigating how DDR pathway exacerbation and resulting cytosolic DNA formation can enhance tumor immunogenicity, activate the innate immune pathway, increase expression of type I interferon (T1IFN) genes, and launch an antitumor immune response. The studies have clear translational implications through informing future clinical trials.
Understanding and overcoming mechanisms of resistance to immunotherapy and chemotherapy
Preliminary data from our group and others suggests that SCLCs evade anti-tumor immunity through repressing major histocompatibility complex class I (MHC class I) expression and antigen presentation. Recently published work suggested that polycomb repressive complex 2 (PRC2) transcriptionally silences the MHC Class I in multiple cancers. We demonstrated that, in SCLC, targeting epigenetic modifiers, like histone demethylase LSD1 restores MHC1 expression and thus anti-tumor immunity. One vital unanswered question is how the immune milieu and microenvironment differ between SCLC subtypes and whether there are any subtype specific effects of the epigenetic modifier targeting in SCLC.
Current projects in the lab include (1) discovering the epigenetic, transcriptional, and immune microenvironmental changes in clinical samples from patients with SCLC; (2) determine the strategy of reactivation of antigen presentation to overcome immune checkpoint blockade resistance in SCLC.
Identification of Novel Targets
There are currently no targeted therapies approved for SCLC patients beyond the recent approval of immunotherapy, which only benefits a small percentage of patients. Drug resistance, both inherent and acquired, is a major problem preventing effective and durable SCLC treatment.
We are developing and employing largescale CRISPR-based pharmacogenomic screens (both in vitro and in vivo) to identify novel drug targets and characterize mechanisms of therapeutic resistance or sensitivity in SCLC.
Non-Small Cell Lung Cancer (NSCLC)
Defining the distinct biology and therapeutic vulnerabilities of KRAS-mutant NSCLC with co-occurring genomic alterations
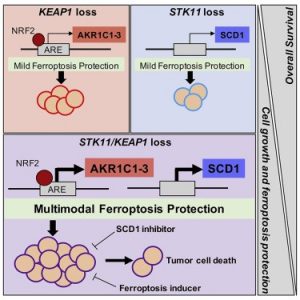
New therapeutic strategies are urgently needed to improve outcomes for patients with STK11/KEAP1 co-mutant non-small cell lung cancer (NSCLC). Concurrent STK11/KEAP1 loss-of-function (LOF) predicts poor (median overall survival of only 7.3 months) prognosis in NSCLC; compared to wildtype or single mutants. STK11/KEAP1 LOF mutations dramatically enhance cell proliferation, invasive potential in vitro, and tumor growth in xenograft lung cancer models. Furthermore, NSCLC cell lines harboring single or co-mutation of STK11 and KEAP1, showed enrichment of pathways suppressing ferroptosis, and co-mutant cells were resistant to ferroptosis-mediated death. CRISPR/Cas9-based screening of these cells identified synthetic lethal interactions specific to the co-mutant state, including multiple regulators of ferroptosis. Our data suggest that isolated STK11 or KEAP1 mutations uniquely support global metabolic rewiring, ferroptosis evasion and provide multimodal ferroptosis protection in co-mutant models.
Current projects include (1) deeper biological understanding of the aggressive co-mutant subsets of KRAS-mutant NSCLC; (2) establish ferroptosis as a therapeutic vulnerability in STK11/KEAP1 co-mutant KRAS-mutant NSCLC; (3) develop novel therapies for STK11/KEAP1 co-mutant KRAS-mutant NSCLC.
Mechanisms of acquired drug resistance to targeted therapies
Lung cancers driven by driver oncogenes (like EGFR mutations and ALK-translocations) are exquisitely sensitive to small molecule tyrosine kinase inhibitors, however drug resistance invariably develops leading to disease relapse. We are employing CRISPR-based pharmacogenomic screens to identify and characterize mechanisms of acquired resistance to targeted therapies that target EGFR, ALK and many other oncogenic drivers. We will validate the targets and mechanisms through functional studies on cell lines, PDX models and organoids.
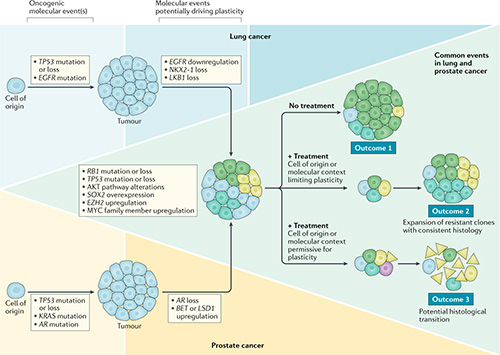
Impact of lineage plasticity on drug response and resistance
Lineage plasticity (LP) is the ability of a tumor to transform from one histologic morphology to another and comprises a central mechanism of acquired resistance. LP was first recognized when an EGFR-mutant LUAD transformed to an aggressive neuroendocrine (NE) carcinoma resembling SCLC. The same phenomenon of NE transformation has been subsequently reported in the context of multiple other oncogenic driver mutations in LUAD, and in multiple other tumor types, including breast and prostate adenocarcinomas. We have previously observed that NE transformation in LUAD (with or without EGFR mutation) enhances therapeutic resistance and metastatic potential. Derivative therapy-resistant transformed-SCLC (T-SCLC) tumors typically exhibit molecular and clinical characteristics similar to de novo SCLC, including a high prevalence of RB1 and TP53 alterations, expression of NE markers, and transient response to platinum-based chemotherapy. Our data suggest that RB1 and TP53 mutations are required, but not sufficient, for NE-transformation. Understanding these mechanisms and developing effective strategies to prevent histologic transformation in lung cancer are critically important.
The focus of our lab is to systematically uncover core mechanisms facilitating lung cancer plasticity in a driver mutation agnostic manner. We are applying single cell technologies and gene editing techniques in clinical tumor samples patient-derived models during therapy to discover the cell states of tumor cells during LP. We aim to identify tumor cell intrinsic and immune microenvironmental factors contributing to LP. Our long-term goal is to develop therapies to reverse or prevent LP as a mechanism of acquired resistance.
Biology and therapies of thoracic neuroendocrine tumors
Neuroendocrine tumor (NET) is a diverse, heterogeneous tumor with a wide range of malignancy, developing tissue, differentiation, and functional status. Among them, thoracic NET is the most favorable site and exhibits a variety of tumor types, from carcinoids with slow growth to aggressive such as small cell lung cancer (SCLC).
Also, while the frequency of total lung cancer occurrence is decreasing, that of NET is increasing due to increasing access and availability of imaging modalities such as computed tomography, indicating that there may be more opportunities to bother us. Therefore, an improved understanding of this group of diseases is a critical need. Also, due to the rarity of well-differentiated lung neuroendocrine tumors, overall progress has been still slow in the development of novel therapeutic clinical trials.
Our research is focused on a deeper understanding of the genomic and transcriptomic characteristics to help predict different therapeutic activity. Our long-term goal is to develop newer therapeutic strategies that can deliver improved outcomes.
Building a preclinical platform for testing novel therapies
We have established an integrated preclinical platform for generation of patient-derived xenograft (PDX) models from tumor or blood samples. These models are a powerful resource for conducting co-clinical trials, identifying mechanisms of resistance, testing the effectiveness of new drugs and finally aid the selection of the most promising drug for evaluation in clinical trials.
FUNDING
We are grateful to the following sponsors for their support:
FUNDING
We are grateful to the following sponsors for their support:







Ongoing Grants:
NIH R01:
Defining and overcoming lineage plasticity in lung cancer.
Debiopharm grant:
Targeting DNA damage response via WEE1 inhibition (Debio123) to enhance anti-tumor efficacy of immunotherapy in small cell lung cancer.
Contact Us
Principal Investigator
Triparna Sen, PhD
Associate Professor
Co-Director, Lung Cancer PDX Program
E-mail
triparna.sen@mssm.edu
Lab Address
Icahn School of Medicine at Mount Sinai
1425 Madison Avenue, 15th Floor
New York, NY 10029
United States
Office Address
1425 Madison Avenue, 15th Floor
New York, NY 10029
United States