|
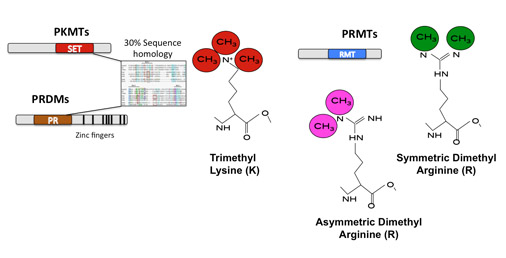
Methyltransferases in Development and Disease. Our group has a long-term interest in the function of Methyltransferases in Development and Disease. In the lab we use a combination of approaches to study the function of Protein Arginine Methyltransferases (PRMTs) and PRDI-BF1-RIZ-homology-domain proteins (PRDMs) (Fig.1) in transcriptional and post-transcriptional regulation during development and cancer: Functional screens and Target Identification and Validation: Conditional Transgenic Knockout (KO) mice: Once we identify direct targets for intervention, we next aim to understand the effect of their depletion in vivo during development and tumorigenesis. To study the former, several projects in the lab have taken advantage of lineage-specific deleter-strains to study deletions of selected methyltransferases in different tissues (Testis and Brain among others). For cancer related projects we mainly take advantage of tumor-prone models such as the Eμ-Myc lymphoma model (Schmitt et al., 1999), which overexpresses the oncogenic transcription factor Myc (Guccione et al., 2006) (Martinato et al., 2008) (Guccione et al., 2007). Biochemistry: All our projects aim at dissecting the biochemical mechanisms driving the in vivo phenotypes we observe upon depletion of the selected methyltransferases. We use in vitro methylation-assays, cell fractionation, in vitro protein-protein interactions and SILAC based quantitative Mass Spectrometry (Migliori et al., 2012) to answer a variety of questions. Bioinformatics: a fundamental part of the work done in the lab deals with large datasets deriving from Chip-Sequencing profiles (of Transcription Factors/co-factors or Histone Post Translational Modifications), DNA methylation landscapes or RNA-Sequencing profiles of wild-type or PRMT/PRDM depleted cells or tissues. We also actively collaborate with other groups both internationally and within IMCB, providing our expertise in computational biology (Beillard et al., 2012; Filipponi et al., 2013; Jachowicz et al., 2013; Tsai et al., 2013; Wang et al., 2014). |