
Stahl Lab Projects
We specialize in aggregate analyses of large genetic datasets, to infer genetic architecture with a wide range of applications. Our primary focus is in psychiatric disease including bipolar disorder and schizophrenia, as part of large-scale collaborative projects. We recently have begun another focus on serum urate and gout disease genetics. In addition, we use polygenic analysis to investigate genetic architecture across traits, within and between populations, and in genomic risk prediction.
Bayesian inference of genetic architecture
We developed Approximate Bayesian Polygenic Analysis (ABPA) [Stahl 2012, Ripke 2013], a Bayesian inference procedure based on polygenic score analysis and its simulation under genetic architecture models. ABPA has provided estimates for common variant genetic architecture including SNP-heritability (total phenotype variance explained) and number of independent associations (shown at right), as well as distributions of associated allele frequencies and effect sizes. We have contributed to numerous other aggregative genetic analysis methods and applications. We are currently preparing ABPA for public software release, validating it under a range of model extensions including Gaussian and semi-parametric (multinomial) priors, multiple genomic compartments (annotations or pathways), and multiple phenotypes, and further extending the approach to better handle linkage disquilibrium and to use alternative objective functions.
Rare variant genetic architecture inference has become a recent focus, and we are analyzing a hierarchical Bayesian approach to identify risk genes based on their frequencies of impactful mutations as well as widely ranging external data including gene sets/pathways, expression profiles, annotations, and others. We are developing a framework for the integrative analysis of large-scale sequence data, accounting for common genetic variation and secondary phenotypes, and developing natural genomic predictors for disease risk and related clinical contexts.
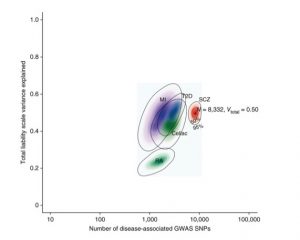
Inferred polygenic architectures of five common diseases
Genetics and Genomics of Psychiatric Disorders
We are engaged in the genome-wide association study of bipolar disorder, as part of the Psychiatric Genomics Consortium Bipolar Disorder Analysis group. Results presented at the 2014 World Congress on Psychiatric Genetics included more than 17,500 BD cases and 26,000 control subjects, and multiple new bipolar disorder associated loci. Bipolar disorder associated loci implicate brain expressed genes with synaptic function, and correlate with loci associated with schizophrenia and other neuropsychiatric traits.
In addition to the primary association analyses, we are working to understand the constituents of bipolar disorder genetic artchitecture in terms of functional DNA elements as well as pathways and sets of genes that function together. We hope to identify and characterize the unique genetic characteristics of bipolar disorder subtypes (BD1, BD2, schizoaffective disorder bipolar type), the similarities and differences between schizophrenia and bipolar, and the neuropsychological traits (mania, positive and negative symptoms, cognitive and social function) that cut across psychiatric disease diagnoses.
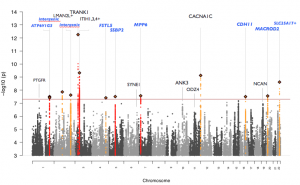
Manhattan plot of PGC_BD Oct 2014 freeze
We lead the analysis of bipolar disorder in whole-exome sequences from the Sweden national hospital registry, which include 1100 bipolar cases, 2500 schizophrenia cases and 2500 controls. Analyses of schizophrenia versus controls were recently published [Pubmed] and are described here. Joint analyses of synaptic gene sets mutated in schizophrenia show the similarity of bipolar to schizophrenia versus controls in rare-variant frequencies. We are working to estimate the rare-variant genetic correlation between bipolar and schizophrenia, which show a high correlation for common genetic variation, and to identify genes that may be specific to bipolar disorder.
We are meta-analyzing the Sweden bipolar disorder whole-exome sequence data with other datasets as part of the case-control (CC) workgroup of the Bipolar Sequencing Consortium. The current data freeze of the BSC_CC group includes 3500 cases and 4700 controls, and will additionally include multi-ethnic samples from Kaiser Permanente and Dutch samples from UCLA. Meta-anlysis of our sequence data will provie greater power to detect genes with excess mutations in bipolar disorder cases, and involves the development of rare-variant meta-analysis methods.
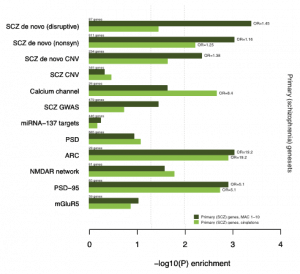
Gene-disruptive mutation enrichments in Sweden exomes
Genetics and Genomics of Urate and Gout Disease
We have recently embarked on a project to conduct multi-ethnic fine-mapping of serum urate loci, by meta-analysis of large-scale GWAS from European samples, Japanese samples, and several multi-ethnic cohorts including our own Mount Sinai Biome biobank. We are also conducting analyses to identify new associated variants in known urate loci, and to infer the most likely causal variants and test them in cellular models. We are funded by an NIH grant for this work, with funding for a postdoctoral researcher position.
With collaborators at Massachusetts General Hospital, Brigham and Women’s Hospital, and the University of Otago in New Zealand, we are also sequencing a 2MB target made up of ~200 implicated genes (coding and regulatory regions), to find rare variants of large effect. We’ve already found high-impact variants in extreme-phenotype samples, and have forwarded them for functional validation in cellular models. Broader analyses of gene and pathway rare variant associations with hyperuricemia are ongoing.
We started this project in search of gout-specific immune/inflammatory genetic factors. We conduct polygenic score-based pathway analyses to dissect the rols of urate metabolism, urate transport and inflammation in gout disease. We are also engaged in other approaches to the genetic epidemiology of gout.
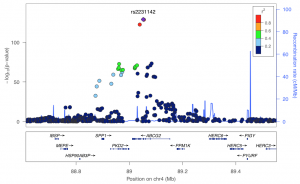
Strong association at a urate transporter locus
Genetic Architecture across Traits and Populations
Data integration for insights from genetic association
We work to interpret genetic association results in the context of genes and pathways that work together in pathogenesis. We have shown enrichment in GWAS of genes connected to each other through PubMed abstracts, of tissue-specifically expressed genes, and of regulatory regions identified through histone Chip-seq experiments including NIH Epigenomics Roadmap. We collaborate within Psychiatric Genomics on methods and neuropsychiatric applicaitons, and Broad Institute Medical and Population Genomics Program members Soumya Raychaudhuri and Ron Do for methods and applications to cardiovascular and autoimmune disease.
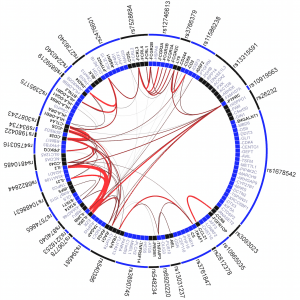
Significant GRAIL connectivity among rheumatoid arthritis loci
Polygenic analysis across traits and populations.
We conduct polygenic analyses across a range of traits and population samples in order to gain insights into their genetic architectures, and to develop long-term prospects for polygenic risk prediction. For example, the variance explained by SNPs ranked by their association evidence has implicaitons for their distribution of effect sizes, and for the population-specificity of their effects. We test how and why genomic predictors based on large-scale genetic studies (primarily of European-descent patients), dissentangling differences in population genetic structure from genetic etiology, and are developing better polygenic risk predicors for non-European populations.
We work in collaboration with the BioMe biobank project and the Institute for Personalized Medicine at Mount Sinai, and the PAGE consortium. The goals of this work are to leverage the rich clinical data existing in electronic health records to discover novel genetic risk factors, and to focus on the ultimate translation of these findings back to the clinic particularly in under-represented populations. This NIH-funded work has an available postdoctoral researcher position.