
1) Identify and characterize the genetic fingerprint of breast cancers (tumor suppressors and oncogenes)
We are in the post-cancer genomics era; an unprecedented amount of information has been gathered through the comprehensive analysis of cancer genomes. However, our ability to analyze this information and identify cancer relevant genes is limited. We have presented a major advance in addressing this challenge using integrative methods, including Helios (Sanchez-Garcia et al. Cell 2014) and ISAR-DEL (Marshall et al. Cancer Cell-under review), that combine genomic data from primary tumors with genome-wide functional studies in cell lines (Figure 1).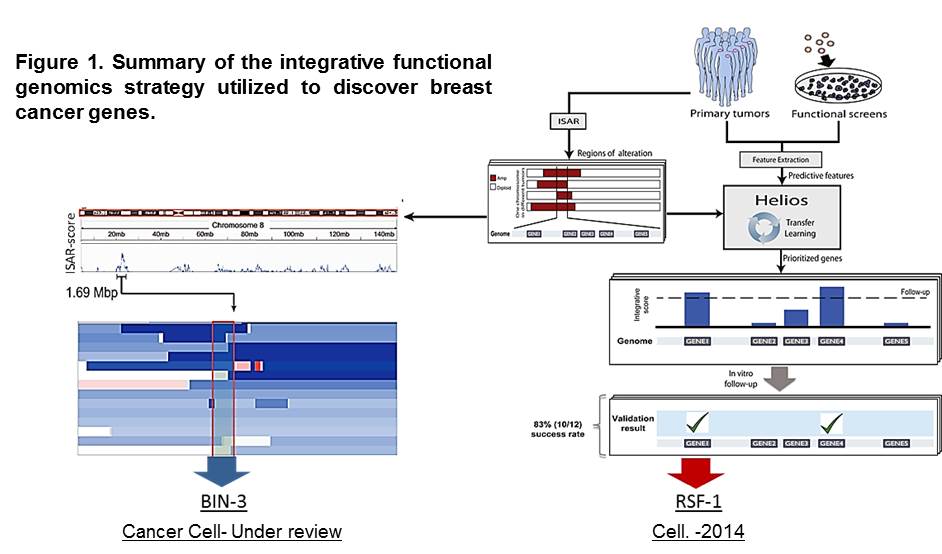
The unparalleled sensitivity and specificity of our approaches enabled us to execute the first reported systematic validation of an algorithm designed to identify driver genes. Our discoveries have significantly expanded the landscape of high-confidence breast cancer drivers by more than 2-fold.
In depth characterization studies revealed RSF-1 as a new breast cancer oncogene with an important role in metastasis (Sanchez-Garcia et al. Cell 2014). RSF1 was found to be amplified and overexpressed in half of the highly aggressive luminal-B subtype of breast cancer tumors. Attenuation of RSF1 compromised tumor cell growth and inhibited metastasis formation. Our new data suggests that RSF1 modulates a stem cell transcriptional program that is required for tumor progression. Furthermore, we have found that co-amplification of RSF1 and the bona fide oncogene CCND1 defines a group of breast cancer patients with reduced survival.
The current and future objectives of this research are to:
- Characterize the stem cell-related transcriptional program modulated by RSF1.
- Study the association of RSF1 and CCND1 in luminal breast cancers.
- Evaluate targeting RSF-1 as an anticancer strategy.
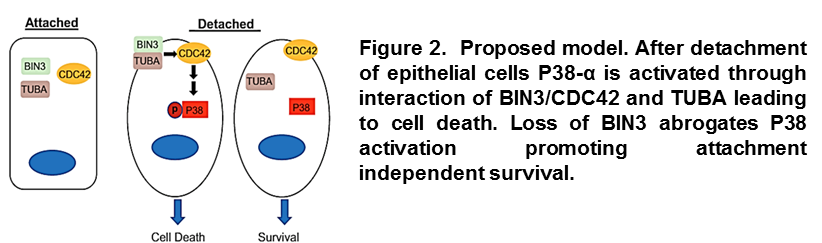
Our studies have also identified BIN3 as a tumor suppressor in the chromosomal region 8p21, which is deleted in almost half of all breast cancers. We demonstrated that loss of expression of BIN3 enhances primary tumor growth and promotes metastasis. Mechanistically, we linked its tumor suppression function to its ability to sense changes in cell membrane structure (cell rounding), which is induced by loss of attachment. After detachment, BIN3 moves to the cell membrane and modulates the relocation and function of CDC42. In these conditions, CDC42 transmits the signal that leads to the activation of the stress protein P38-α and programmed cell death (Figure 2).
Overall, our results explain how changes in cell geometry are integrated in the cellular signaling network and present, for the first time, BIN3 as a novel breast cancer tumor suppressor (Marshall et al. Cancer Cell-under review).
The current and future objectives of this research are to:
- Characterize the regulation of BIN3 in normal and breast cancer cells.
- Evaluate modulation of the signaling defined by BIN3-P38 to reduce the metastatic potential of cancer cells.
2) Genome-wide functional genomics studies to identify novel tumor targets (oncogene and non-oncogene addiction hubs)
Cancer treatment is rapidly evolving toward more efficient and less harmful targeted therapies. We have successfully pioneered the integration of state-of-the-art genomics, functional studies, and system biology interactome models for the reverse engineering of regulatory networks. These analyses pinpoint key regulatory hubs that, when perturbed, compromise cancer cell viability (Rodriguez-Barrueco et al. Genes & Dev. 2015; Putcha et al. Breast Cancer Res. 2015 (Figure 3)).
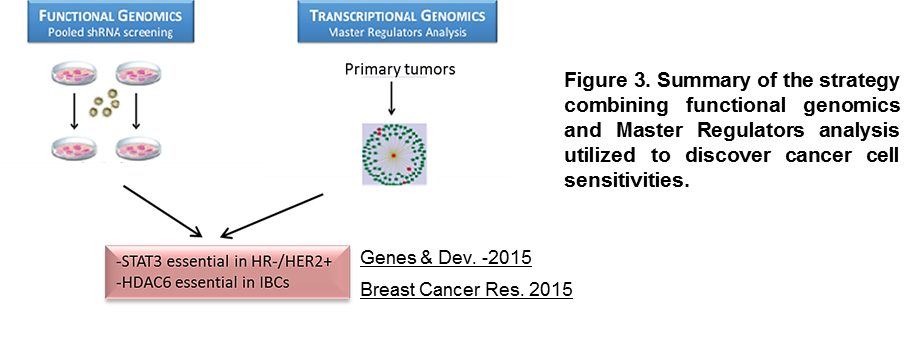 Using this strategy, we have discovered that the JAK/STAT3 pathway is activated in HR-/HER2+ breast tumors and, importantly, it is essential to maintain their homeostasis. Mechanistically, we found that HR-/HER2+ cells secrete high levels of IL-6, generating an autocrine loop which induces activation of STAT3 through the canonical JAK/STAT pathway. Activated STAT3 initiates a transcriptional program that upregulates the S100A8/9 complex (calprotectin). This complex is also secreted and activates a second autocrine loop that leads to higher activation levels of the pro-survival AKT signaling pathway. We demonstrated that inhibition of the IL-6-JAK2-STAT3-Calprotectin axis with three different FDA-approved drugs, alone and in combination with HER2 inhibitors, reduced the tumorigenicity of HR-/HER2+ breast cancers (Figure 4). Our studies have led to a phase I/II, multi-center trial (gov identifier: NCT02066532) (Rodriguez-Barrueco et al. Genes & Dev. 2015).
Using this strategy, we have discovered that the JAK/STAT3 pathway is activated in HR-/HER2+ breast tumors and, importantly, it is essential to maintain their homeostasis. Mechanistically, we found that HR-/HER2+ cells secrete high levels of IL-6, generating an autocrine loop which induces activation of STAT3 through the canonical JAK/STAT pathway. Activated STAT3 initiates a transcriptional program that upregulates the S100A8/9 complex (calprotectin). This complex is also secreted and activates a second autocrine loop that leads to higher activation levels of the pro-survival AKT signaling pathway. We demonstrated that inhibition of the IL-6-JAK2-STAT3-Calprotectin axis with three different FDA-approved drugs, alone and in combination with HER2 inhibitors, reduced the tumorigenicity of HR-/HER2+ breast cancers (Figure 4). Our studies have led to a phase I/II, multi-center trial (gov identifier: NCT02066532) (Rodriguez-Barrueco et al. Genes & Dev. 2015).
The current and future objectives of this research are to:
- Evaluate targeting the IL-6-JAK2-STAT3-Calprotectin axis as an anticancer breast cancer therapy.
- Investigate mechanisms of resistance to inhibitors targeting the IL-6-JAK2-STAT3-Calprotectin axis.
- Investigate the expansion of tumor initiating cells mediated by activation of the IL-6-JAK2-STAT3-Calprotectin axis and its implications for cancer progression and therapeutics.
Inflammatory breast cancer (IBC) is the most lethal form of breast cancer with a 5-year survival rate of only 40%. Despite its lethality, IBC remains poorly understood and management mainly relies on chemotherapy. Through our integrative studies, we have recently found that HDAC6 functions as a “non-oncogene” addiction hub in IBC cells. We showed that HDAC6 activity is significantly higher in IBCs than in non-IBCs, which suggests a mechanism that mediates this dependency. Importantly, small molecule inhibitors for HDAC6 already exist and are in clinical trials for other tumors; we have shown their activity against IBC cells in pre-clinical models (Putcha et al. Breast Cancer Res. 2015).
HDAC6 has emerged as a Master Regulator of the cell’s protective response to accumulation of toxic bioproducts (misfolded proteins and damaged mitochondria) (Figure 5). Currently, we are investigating which of HDAC6’s canonical/non-canonical functions is involved in the sensitivity to HDAC6 inhibition.
Furthermore, we have found that a subset of HR+ non-IBC breast cancers (~30%) also present high activity levels of HDAC6 and that HR+ cells with high HDAC6 activity showed a strong dependency of this function. Our research has led to the development of a new clinical trial that will begin during the last quarter of 2015.

The current and future objectives of this research are to:
- Characterize the molecular mechanism that defines the dependency of IBC cells on HDAC6 activity.
- Investigate the dependency of HR+ non-IBC cells on HDAC6 function.
- Investigate mechanisms of resistance to HDAC6 inhibitors.
3) miRNAs that regulate the development of the mammary epithelium and their contribution to tumorigenesis
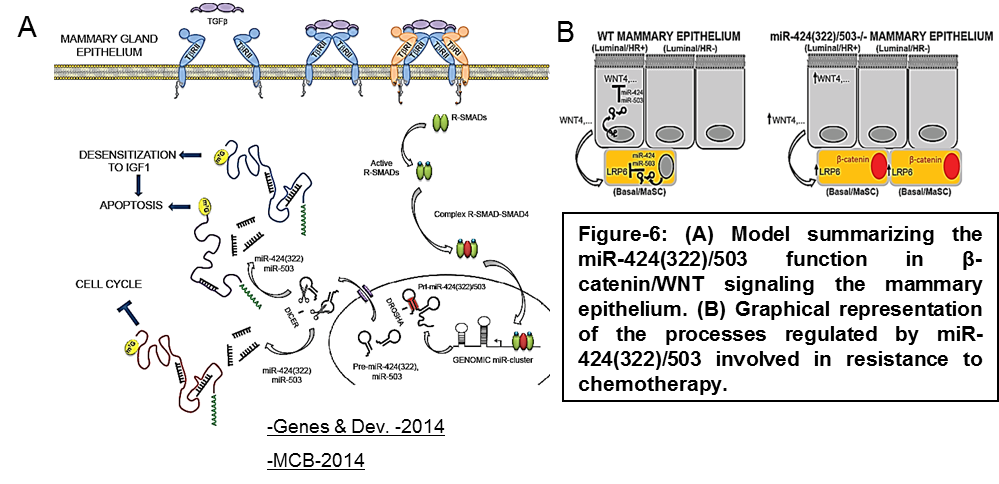
We have identified the miR-424(322)/503 cluster as an important regulator of mammary involution after pregnancy (Llobet-Navas et al. Genes & Dev. 2014; Llobet-Navas et al. MCB 2014). Mechanistically, our studies unveiled that miR-424(322)/503 is induced by the canonical TGF-β-SMAD pathway, and that it orchestrates downregulation of key components of signal transduction, apoptosis, and cell cycle (Figure 6A). Recently, we have also found that miR-424(322)/503 modulates canonical WNT/β-catenin signaling through the control of LRP6 and WNT4 expression (Figure 6B).
Our analysis of over 2,000 primary breast cancers (Metabric) revealed that this miRNA cluster is deleted in about 16% of breast tumors and, consistently, miR-424(322)/503-/- female mice develop hyperplasias and mammary tumors that are promoted by pregnancy. We have also demonstrated that loss of the miR-424(322)/503 cluster promotes resistance to chemotherapy due to its ability to modulate the expression of IGF1R and BCL-2 (Figure 6A) and that the use of IGFR1 and BCL2 inhibitors (already in clinical trials) restores the sensitivity to chemotherapy (Rodriguez-Barrueco et al. Genes & Dev. 2017).
Overall, we have characterized a previously unknown, multilayered regulation of epithelial tissue remodeling after pregnancy that is coordinated by the miR-424(322)/503 cluster. Additionally, we have unveiled its tumor suppressor role and provided therapeutic alternatives for breast cancer patients with deletions in this locus.
The current and future objectives of this research are to:
- Investigate putative targets of miR-424(322)/503 in the mammary epithelium.
- Characterize the alterations of the miR-424(322)/503→targets axis in breast cancers (including pregnancy-associated tumors).