
How a CDK network coordinates the cell division cycle
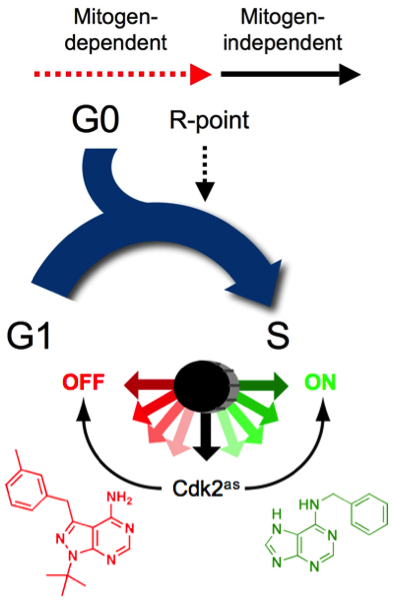
Figure 1. Cdk2, a rate-limiting regulator of cell-cycle commitment. Passage of the R point, when further cell-cycle progression becomes mitogen-independent, is dependent on the activity of Cdk2. We showed this by replacement of wild-type Cdk2 with Cdk2as, which could be switched on with 6-benzylaminopurine (green) or off with 3-methylbenzyl-PP1 (red) to accelerate or impede R-point passage, respectively (see Merrick et al., 2011).
A chemical-genetic window on restriction point regulation
To define the specific functions of a single, essential kinase in human colon cancer-derived cells, we replaced it with an analog-sensitive (AS) mutant version; allele-specific inhibition of Cdk7as revealed essential CDK-activating functions at the entry to DNA synthesis (S) phase and mitosis. We showed that Cdk7 enforces CDK-cyclin pairing specificity, and thus the order in which different CDKs are activated during the cell cycle, by supporting distinct activation pathways for two CDKs that bind the same cyclin. Then, by targeting one of those CDKs, Cdk2, we uncovered its essential functions—to promote commitment to division at the Restriction (R) Point and entry to S phase. The AS mutant version of Cdk2 acted in vivo as a three-position switch: activation by one small molecule accelerated passage of the R point, whereas inhibition by another compound blocked that progression (Figure 1). Therefore, in wild-type cells, Cdk2 is a non-redundant, rate-limiting regulator of cell-cycle commitment.
One activating kinase but distinct activation pathways for different CDKs
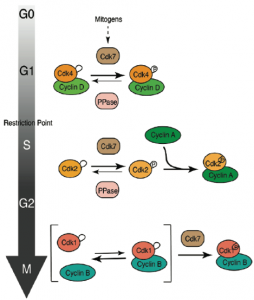
Figure 2. Changing modes of CDK activation during cell-cycle progression: from analog sensor to binary switch. Although they share a common CAK (Cdk7), the CDKs active during G1, S/G2 and M phases follow different kinetic paths to activation, which contributes to CDK-cyclin pairing specificity and the regulation of cell-cycle commitment by mitogen signaling (see Larochelle et al., 2007; Merrick et al., 2008; Schachter et al., 2013).
The ability to inhibit Cdk7 selectively allowed us to show that it is the activating kinase for Cdk2, the major CDK active during S and G2 phases, and for Cdk1, which triggers mitosis. It remained unclear what role Cdk7 played during early G1, when Cdk4 and Cdk6 are active, or whether these cyclin D-dependent kinases might depend on a different CAK to be activated in vivo. A clue emerged when we compared the effects of Cdk7 versus Cdk2 inhibition in Cdk7as or Cdk2as cells, respectively; Cdk7 inhibition was more effective in blocking S-phase entry, suggesting it had targets besides Cdk2 required for G1/S progression. Indeed, Cdk7 inhibition rapidly shuts off activation of both Cdk4 and Cdk6, and prevents them from phosphorylating the retinoblastoma tumor suppressor protein pRB to initiate transcription of cell-cycle genes. We showed that the activation state of Cdk7 itself is regulated by mitogens, and that a Cdk7-Cdk4/6 phosphorylation cascade might be rate-determining for R-point passage. Thus, CDK activation in human cells depends on a common CAK, Cdk7, which can support distinct pathways of activation at different points in the cell cycle (Figure 2). This raises the possibility that pathway-specific inhibition of CAK-CDK interactions might act to block cell-cycle progression at different points in the cycle. Current aims are to define signaling pathways that promote Cdk7 activation during G1, and to map synthetic-lethal interactions with CDK inhibitors.
This work is supported by NIH grant RO1-GM105773, “Chemical Genetic Analysis of the Human Cell Cycle.”
A transcription cycle driven by CDKs?
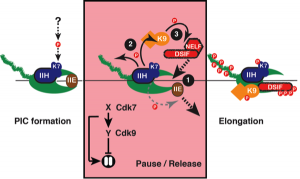
Figure 3. A CDK-dependent switch in transcription. Chemical genetics uncovered two requirements for TFIIH-associated Cdk7 (K7) in the transition from preinitiation complex (PIC) to elongating transcription complex: 1) recruitment of elongation factors DSIF and NELF and displacement of initiation factor TFIIE to establish a paused complex; and 2) activation of Cdk9 (K9) to release RNAP II from the pause (see Larochelle et al., 2012).
The involvement of CDKs in regulating the transcription cycle of RNAP II is conserved throughout eukaryotic evolution, but the precise functions and targets of these transcriptional CDKs remain largely unknown. By the chemical-genetic strategy—replacement of wild-type Cdk7 with Cdk7as in human cells—we defined requirements for Cdk7 to govern the initiation-elongation transition of RNAPII. This is an ancient point of transcription regulation, conserved even in archaea and prokaryotes, in which RNAP is “handed off” from initiation to elongation factors. Our studies suggest that, in eukaryotes, this handoff has come under the control of CDKs. Cdk7 activity is required to establish the promoter-proximal pause in RNAPII elongation, and to activate the Cdk9/cyclin T1 complex, also known as positive transcription elongation factor b (P-TEFb), which releases the pause (Figure 3). This incoherent feedforward loop may work to ensure promoter-proximal recruitment of RNA-processing and chromatin-modifying machinery.
We showed that the fission yeast orthologs of TFIIH and P-TEFb also act in defined sequence to coordinate recruitment of mRNA-processing and chromatin-modifying enzymes. Thus the strict ordering of discrete CDK functions is conserved between yeast and human cells, and between transcription and cell-division cycles. By analogy with the cell cycle, a full understanding of the transcription cycle will require characterization of each CDK involved, and its protein substrates. Current aims are to determine the mechanism of Cdk7-dependent promoter-proximal pausing, characterize genetic and biochemical interactions between transcriptional CDKs and histone H2B mono-ubiquitylation—a conserved chromatin modification implicated in transcript elongation—and identify transcriptional proteins phosphorylated by Cdk7 and/ or Cdk9 in yeast and human cells. We hope to uncover novel functions of the CDK network in RNA quality control, which might be targeted with small molecules in human cancer.
This work is supported by NIH grant “R35 GM127289 – Cyclin-dependent kinase control of cell-division and transcription cycles”