
The Structural and Chemical Biology of Cancer Signaling Pathways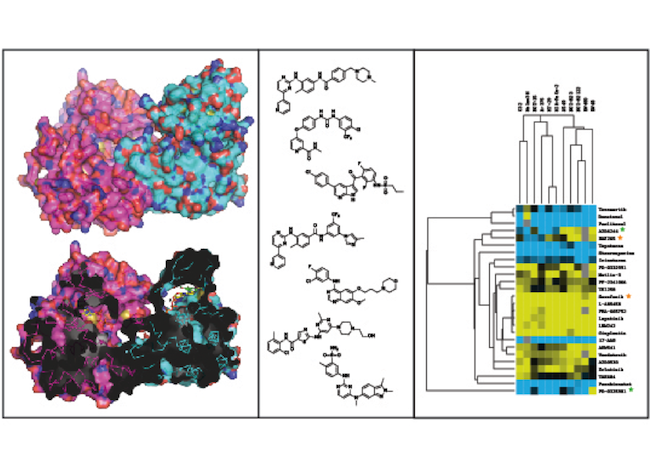
How do the pathways that can drive cancers operate normally? How do mutations found in disease alter the structural and biochemical properties of the pathways? And ultimately how can we selectively disable these pathways when they cause disease? This last question is a major challenge in developing therapeutic approaches for the treatment of cancer, and in large part is what we think about most. Along these lines, we focus efforts on the structural and biochemical properties of the Ras-Mitogen Activated Protein Kinase (MAPK) Pathway. Multicellular organisms use the Ras-MAPK pathway for sensing growth factors and relaying the signals that allow cells to survive and proliferate. Cancer cells are commonly mutated at the level of Ras and its immediate effector RAF. Ras is a GTPase whereas RAF functions as a kinase and when active, these two proteins interact directly with one another. Cancers that contain mutations in Ras or RAF are amongst the most aggressive and difficult to treat human cancers, including pancreatic, colorectal, and lung. We are exploring new stragtegies to modulate the Ras-MAPK based on targets that we have identified through genetic studies and known intrinsic or acquired mechanisms of resistance to current therapeutic approaches. If successful in translating any of our approaches to the clinic, we could impact as many as 1 in 4 of all cancer patients. In the US alone, this would represent over 100,000 patients per year.
Rational Polypharmacology
All clinical drugs share a common feature: the dose at which they are effective is below the dose at which they are toxic. Too low and a drug will not show any efficacy; too high and the side effects of a drug dominate leading to adverse events. Given this paradigm, what if we could identify all targets that contribute to the efficacy of a drug – could we add other targets, making a lead compound much more efficacious at a lower dose? Alternatively, what if we could identify any targets that contribute to the dose-limiting toxicity of a drug? Could we eliminate the activity of a drug against that target and make it safer at much higher levels?
Understanding how a drug inhibits one or many targets within the context of highly interconnected signaling networks can lead to the development of safe and effective therapeutics. Kinases are a large superfamily of enzymes that function as fundamental regulators of biochemical information flow and kinase-targeted drugs have produced several breakthrough therapies for many cancers including chronic mylegenous leukemia and melanoma. However, owing to the degenerate nature of the ATP-binding pocket across all kinases and because most clinically approved kinase drugs are ATP-competitive small molecules, there is often a high probability for multi-targeted activity across the kinase family. There is mounting evidence that most, if not all, small molecule kinase inhibitors derive their efficacy from multi-targeted inhibition of several distinct kinases. Yet many of these targets remain unknown or poorly characterized.
For us to derive the safest, most effective kinase-targeted drugs, not only do we want to inhibit the drivers of disease, eliminate activity against unnecessary targets to reduce side effects, there is also a third consideration. We believe that there are kinases where when activated, can act in a synergistic fashion to enhance the efficacy of a treatment. We call these kinases ‘anti-targets’. Kinase anti-targets have been largely ignored in drug discovery pipelines but they may be highly relevant to the therapeutic efficacy of kinase drugs. We are using multidisciplinary approaches, including chemical and computational biology with genetic screens to identify targets and anti-targets of kinase inhibitors and we use this information to guide the design and synthesis of molecules with improved biological properties. We conduct this work in close collaboration with Drs. Avner Schlessinger and Ross Cagan, both of whom are located here at Mount Sinai.