
B cell defects and Common Variable Immune Deficiency
One of the main projects in the laboratory is the study of a relatively common primary immune deficiency, common variable immune deficiency (CVID). The diagnosis of this defects is made when there are reduced serum levels of IgG, IgA and/or IgM (at least two standard deviations below the mean for age) with reduced or absent antibody production, but with not other medical cause of this profound humoral defect. As for the other immune defects, CVID is associated with an increased number of infections, most often bacterial respiratory tract infections. However, as in other immune deficiency states, inflammatory complications occur in varying proportions and include autoimmunity, chronic lung disease, bronchiectasis, gastrointestinal disease with or without malabsorption, systemic or localized granulomatous disease, liver disease, splenomegaly, lymphadenopathy with or without lymphoma and other malignancies Although the diagnosis is most typically made between age 20 and 40 years of age, about 20% of patients are less than 21 years old at diagnosis, and others give a history of clinical events suggestive of an immune disorder in early childhood. CVID has an estimated prevalence of 1 : 25 000 1 : 50 000, and due to the need for immune globulin treatment, and also because of the large numbers of medical encounters required to treat its associated complications it is a clinically important immune defect. Studies of CVID have included both clinical and laboratory approaches. Some of the current concepts and projects are described here.
Toll like receptors (TLR) and human B cells
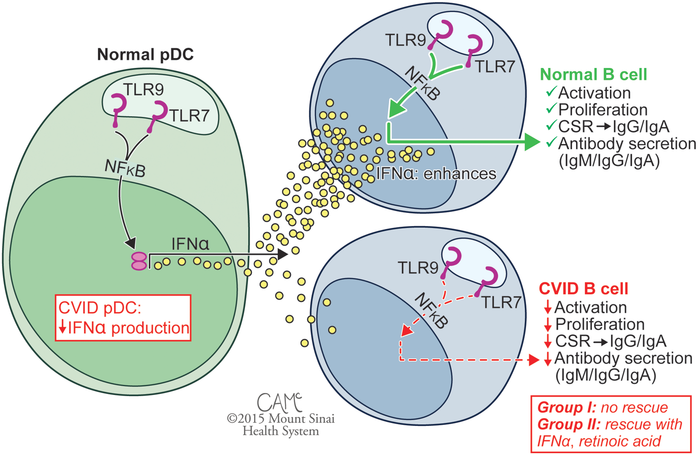
A theme in this Laboratory has been how toll like receptors (TLR) activate the B cell system. B cells are normally activated by nucleic acid-sensing TLR7 and TLR9 receptors, leading to B cell proliferation immune globulin secretion. Our group first reported defective B cell proliferation and Ig secretion in response to CpG motif–containing nucleotides in CVID, although the mechanism was unknown as this preceded the discovery of mammalian TLR9. Following the characterization of human TLR9 and the discovery of powerful synthetic agonists, our group and others revisited responses of CVID patients to TLR9 stimulation, finding pervasive B cell defects in activation and in cytokine secretion when, notably, patients did not have mutations or variations in TLR9. While the normal pDC response to stimulation with TLR7 and TLR9 agonists is secretion of high levels of IFN-α, pDCs of subjects with CVID show defective responses. Similarly, while normal B cells are sensitive to stimulation with TLR7 and TLR9 agonists, patient B cells show defective responses. The B cell defect is correctable in less severely affected Group II patients with the addition of exogenous IFN-α or retinoic acid, but these fail to rescue B cells from Group I patients.
Figure 1: Patients are subdivided into Group I (<0.55% SWMB, associated with noninfectious complications) and less severely affected Group II (>0.55% SWMB, associated with better clinical course). CVID, common variable immunodeficiency; SWMB, class switched memory B cells. Figure courtesy of Courtney A. McKenna © 2015 Mount Sinai Health System.
T cell defects in CVID:
While the Laboratory has often investigated impaired B cell immunity in CVID, numerous functional defects of T cell immunity have been described by ourselves and others over the past several decades. In a new study, we used high throughput sequencing to examine the structure and composition of the β chain of the CVID T cell receptor. We examined junctional diversity, extent of mutational changes, and degree of clonality to assess the repertoire of CVID T cells. TCRb CDR3 regions were amplified and sequenced from genomic DNA of 44 adult CVID subjects and 22 healthy adults, using a high-throughput multiplex PCR for re-arranged DNA of T cells. Nucleotide sequence, V, D, and J gene usage, copy number, and predicted amino acid sequence were determined. As compared to control DNA, both in-frame and out-of-frame CVID T cell receptors were more germline, had significantly less junctional diversity, fewer n-nucleotide insertions and deletions, and completely lacked a population of highly modified TCRs seen in healthy controls. The CVID CDR3 sequences were significantly more clonal than control DNA, and displayed unique V gene region usage. Despite both the more germline and clonal repertoire and similar infectious exposures, DNA of CVID subjects shared fewer TCR sequences as compared to controls. The expanded germline repertoire, loss of junctional diversity, and clonal expansions in CVID suggests intrinsic T cell defects originating at the thymic level. These abnormalities are pervasive, found in out-of-frame sequences, and thus independent of selection and were not associated with specific clinical complications. These data support an inherent T cell defect in CVID, and provide evidence of a combined immune defect.
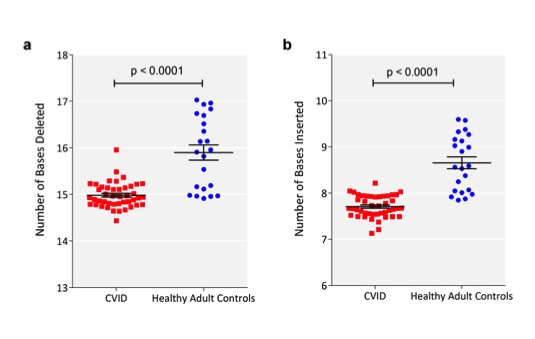 Figure 3 There were fewer VDJ deletions and n-nucleotide insertions in CVID CDR3 sequences compared to normal controls. a) The sum of the numbers of deletions from Vβ, Dβ, and Jβ sequences were calculated for each sequence. The mean number of deletions (in bases) for each patient (red square) or control (blue filled circle) is shown (y-axis). The horizontal bar represents the mean of each group and the whiskers represent the standard error of the mean. b) The sum of the number of insertions between Vβ and Dβ, and Dβ and Jβ sequences were similarly calculated for each sequence. p-values of t-test are indicated.
Figure 3 There were fewer VDJ deletions and n-nucleotide insertions in CVID CDR3 sequences compared to normal controls. a) The sum of the numbers of deletions from Vβ, Dβ, and Jβ sequences were calculated for each sequence. The mean number of deletions (in bases) for each patient (red square) or control (blue filled circle) is shown (y-axis). The horizontal bar represents the mean of each group and the whiskers represent the standard error of the mean. b) The sum of the number of insertions between Vβ and Dβ, and Dβ and Jβ sequences were similarly calculated for each sequence. p-values of t-test are indicated.