
The Marazzi Laboratory (Laboratory of Cellular Response in Health and Disease) studies epigenetic and chromatin-mediated control of gene expression in the context of the cellular response to pathogens or differentiation. We are interested in mechanisms that control the cell’s response. We use biochemistry, genetics, and next generation sequencing techniques to understand molecular mechanisms and genome-wide effects of known and novel candidate genes.
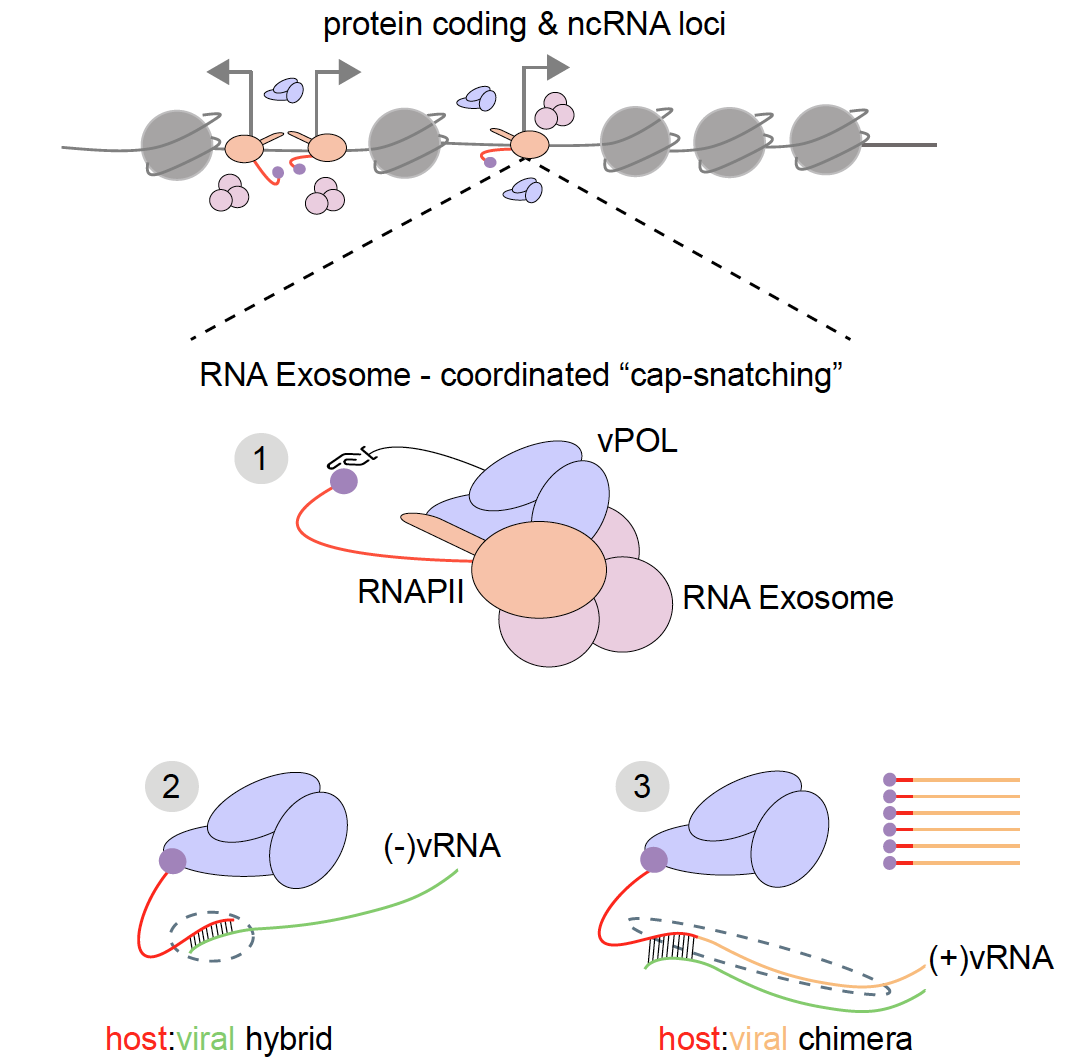
RNA Exosome – Influenza A RdRP Co-option

Inhibiting Topoisomerase I to suppress pro-inflammatory transcription
The host innate immune response is the first line of defense against pathogens and is orchestrated by the concerted expression of genes induced by microbial stimuli. Deregulated expression of these genes is linked to the initiation and progression of diseases associated with exacerbated inflammation. We identified topoisomerase 1 (Top1) as a positive regulator of RNA polymerase II transcriptional activity at pathogen-induced genes. Depletion or chemical inhibition of Top1 suppresses the host response against influenza and Ebola viruses as well as bacterial products. Therapeutic pharmacological inhibition of Top1 protected mice from death in experimental models of lethal inflammation. Our results indicate that Top1 inhibition could be used as therapy against life-threatening infections characterized by an acutely exacerbated immune response.
Unwinding inducible gene expression. Pope & Medzhitov, Science. 2016
Targeting Viral Proteostasis

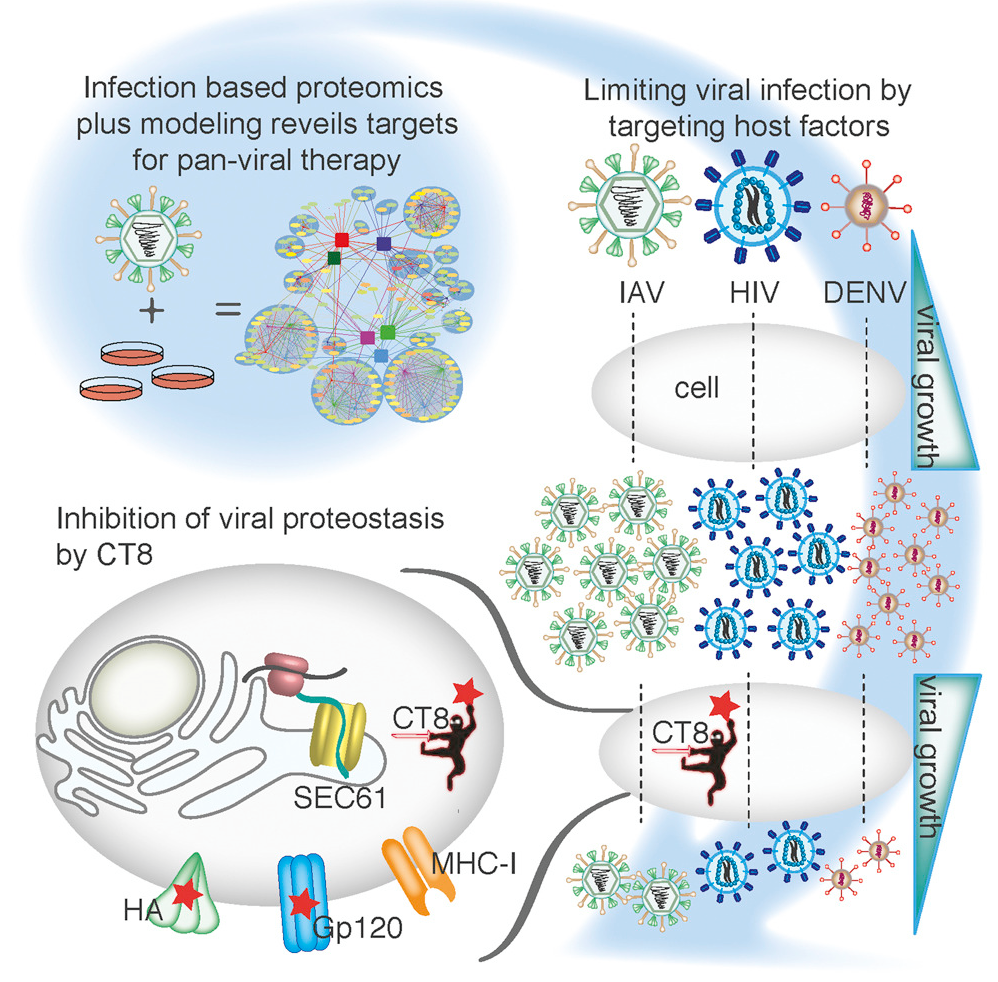
Viruses are obligate parasites and thus require the machinery of the host cell to replicate. Inhibition of host factors co-opted during active infection is a strategy hosts use to suppress viral replication and a potential pan-antiviral therapy. To define the cellular proteins and processes required for a virus during infection is thus crucial to understanding the mechanisms of virally induced disease. In this report, we generated fully infectious tagged influenza viruses and used infection-based proteomics to identify pivotal arms of cellular signaling required for influenza virus growth and infectivity. Using mathematical modeling and genetic and pharmacologic approaches, we revealed that modulation of Sec61-mediated cotranslational translocation selectively impaired glycoprotein proteostasis of influenza as well as HIV and dengue viruses and led to inhibition of viral growth and infectivity. Thus, by studying virus-human protein-protein interactions in the context of active replication, we have identified targetable host factors for broad-spectrum antiviral therapies.
Transcription, Viral Infection, and Neurological Disease
The human helicase senataxin (SETX) has been linked to the neurodegenerative diseases amyotrophic lateral sclerosis (ALS4) and ataxia with oculomotor apraxia (AOA2). Here we identified a role for SETX in controlling the antiviral response. Cells that had undergone depletion of SETX and SETX-deficient cells derived from patients with AOA2 had higher expression of antiviral mediators in response to infection than did wild-type cells. Mechanistically, we propose a model whereby SETX attenuates the activity of RNA polymerase II (RNAPII) at genes stimulated after a virus is sensed and thus controls the magnitude of the host response to pathogens and the biogenesis of various RNA viruses (e.g., influenza A virus and West Nile virus). Our data indicate a potentially causal link among inborn errors in SETX, susceptibility to infection and the development of neurologic disorders.
Viral Mimics
Viral infection is commonly associated with virus-driven hijacking of host proteins. Here we describe a novel mechanism by which influenza virus affects host cells through the interaction of influenza non-structural protein 1 (NS1) with the infected cell epigenome. We show that the NS1 protein of influenza A H3N2 subtype possesses a histone-like sequence (histone mimic) that is used by the virus to target the human PAF1 transcription elongation complex (hPAF1C). We demonstrate that binding of NS1 to hPAF1C depends on the NS1 histone mimic and results in suppression of hPAF1C-mediated transcriptional elongation. Furthermore, human PAF1 has a crucial role in the antiviral response. Loss of hPAF1C binding by NS1 attenuates influenza infection, whereas hPAF1C deficiency reduces antiviral gene expression and renders cells more susceptible to viruses. We propose that the histone mimic in NS1 enables the influenza virus to affect inducible gene expression selectively, thus contributing to suppression of the antiviral response.