

When we have a meal the concentration of glucose goes up in the blood. Cells in the body, including liver cells and pancreatic beta cells, perceive this and respond in a physiologically appropriate manner. In the pancreas, specialized cells called beta cells detect increased glucose and secrete insulin. Liver cells also are aware of increased glucose, and along with insulin, glucose signals liver cells to change their phenotype from the fasting state (with gluconeogenesis, glycogenolysis, and ketogenesis to make glucose and ketones during a period of fasting) to the fed state (with glycogen synthesis and fatty acid synthesis). Normally this switch in phenotype happens reciprocally, so the fasted state is switched off when the fed state is switched on and vice versa. However, in diabetes, the fasted state stays on causing hyperglycemia – and in other metabolic disorders, the switch is incomplete and the fasting state and the fed state are on at the same time resulting in fatty liver disease. In order to change phenotype, cells must alter gene expression patterns, a job performed by transcription factors.
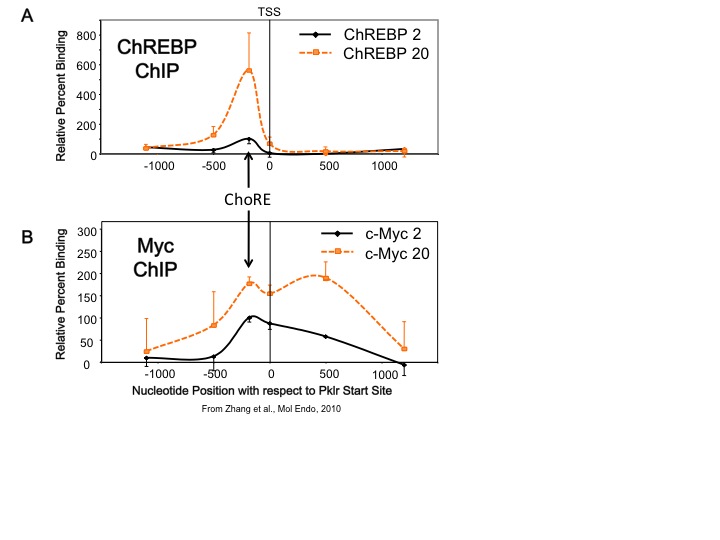
Our lab studies transcription factors whose activities are altered by changes in glucose concentration. One transcription factor is carbohydrate response element binding protein, or ChREBP. This factor is expressed in both liver and pancreatic beta cells. ChREBP is activated by increased glucose metabolism, and so is a transcriptional sensor of blood glucose levels. We found that another transcription factor, Myc, is required for ChREBP to bind to DNA and is required for glucose-mediated gene transcription in both liver and pancreatic beta cells. In addition, we found that Myc is recruited to glucose responsive gene promoters, but it appears that Myc does not require a specific DNA biding site as ChREBP does, but rather that Myc is recruited over a broad region that includes the carbohydrate response element (ChoRE), the transcriptional start site (TSS), and approximately 500 bp downstream of the start site (see Figure). This suggests a new paradigm for how Myc functions to allow cells to change their phenotype in response to altered metabolic environments. Based on these results, we have hypothesized that Myc is required for the natural transition between the fasted to the fed state in the liver.
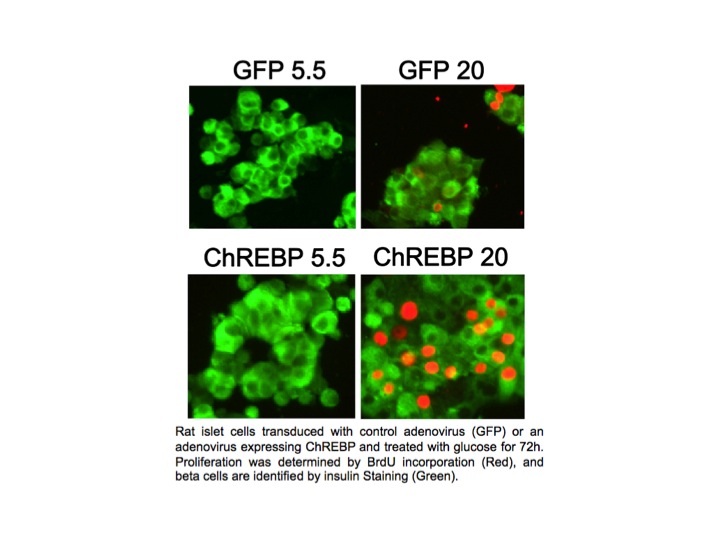
 In pancreatic beta cells, under the right circumstances, increased glucose metabolism stimulates proliferation as part of a homeostatic mechanism whereby beta cell mass expands to accommodate the need for more insulin production. Type I and Type II diabetes are disorders of insufficient beta cell mass, and so an important therapeutic goal is to increase beta cell mass by coaxing beta cells to divide, or proliferate. We found that overexpression of ChREBP amplifies glucose-stimulated beta cell proliferation (see Figure). We hope to understand and manipulate this natural process to the benefit of people with diabetes.
In pancreatic beta cells, under the right circumstances, increased glucose metabolism stimulates proliferation as part of a homeostatic mechanism whereby beta cell mass expands to accommodate the need for more insulin production. Type I and Type II diabetes are disorders of insufficient beta cell mass, and so an important therapeutic goal is to increase beta cell mass by coaxing beta cells to divide, or proliferate. We found that overexpression of ChREBP amplifies glucose-stimulated beta cell proliferation (see Figure). We hope to understand and manipulate this natural process to the benefit of people with diabetes.
We use a variety of methodologies to understand the coordinated regulation of gene expression by glucose, including in vivo and in vitro adenovirus gene delivery, transgenic animals, chromatin immunoprecipitation, nuclear run-ons, real time RT-PCR, Western blots and several variations of gene arrays. A better understanding of these coordinating transcription factors, how these factors interact with and their role in glucose homeostasis and immunological wellbeing will provide new opportunities for designing novel therapies for the treatment of metabolic disorders such as diabetes and obesity.